

Multiscale Methods in Drug Design Bridge Chemical and Biological Complexity in the Search for Cures
- PMID: 30949587
- PMCID: PMC6445369
- DOI: 10.1038/s41570-018-0148
Multiscale Methods in Drug Design Bridge Chemical and Biological Complexity in the Search for Cures
Abstract
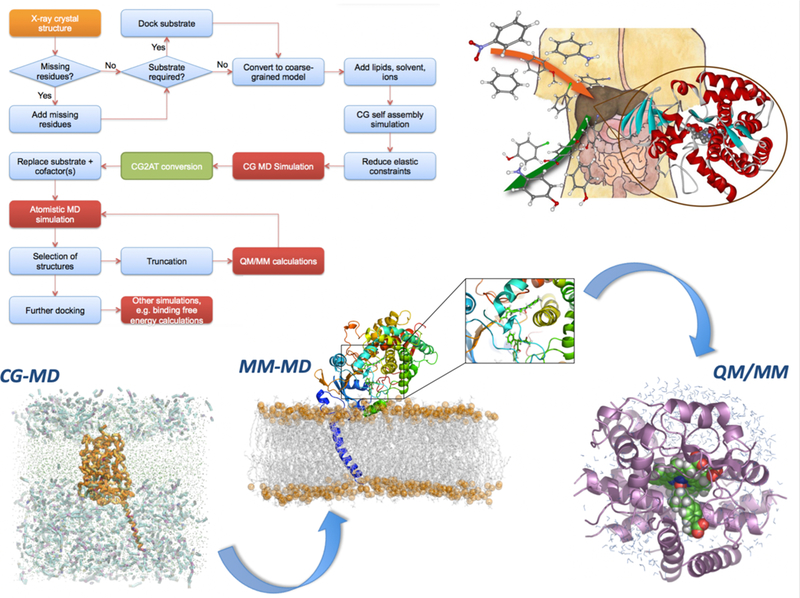
Drug action is inherently multiscale: it connects molecular interactions to emergent properties at cellular and larger scales. Simulation techniques at each of these different scales are already central to drug design and development, but methods capable of connecting across these scales will extend understanding of complex mechanisms and the ability to predict biological effects. Improved algorithms, ever-more-powerful computing architectures and the accelerating growth of rich datasets are driving advances in multiscale modeling methods capable of bridging chemical and biological complexity from the atom to the cell. Particularly exciting is the development of highly detailed, structure-based, physical simulations of biochemical systems, which are now able to access experimentally relevant timescales for large systems and, at the same time, achieve unprecedented accuracy. In this Perspective, we discuss how emerging data-rich, physics-based multiscale approaches are of the cusp of realizing long-promised impact in the discovery, design and development of novel therapeutics. We highlight emerging methods and applications in this growing field, and outline how different scales can be combined in practical modelling and simulation strategies.
Figures

References
Grants and funding
LinkOut - more resources
Full Text Sources
