

Carbonic Anhydrase Inhibition Ameliorates Inflammation and Experimental Pulmonary Hypertension
- PMID: 30951642
- PMCID: PMC6775956
- DOI: 10.1165/rcmb.2018-0232OC
Carbonic Anhydrase Inhibition Ameliorates Inflammation and Experimental Pulmonary Hypertension
Abstract
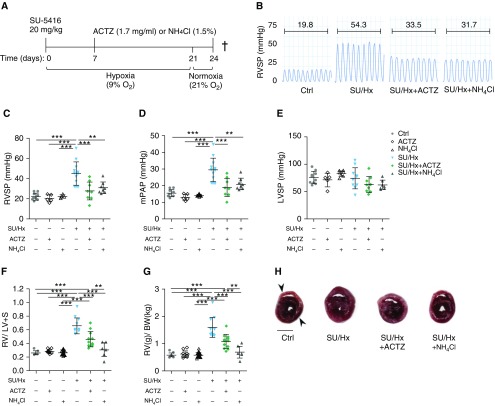
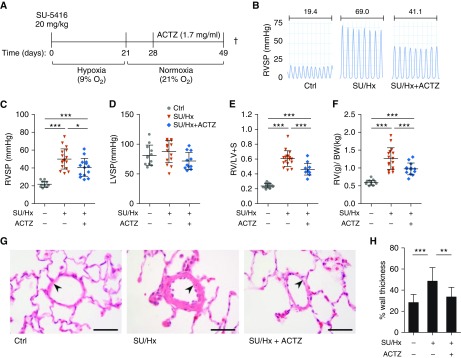
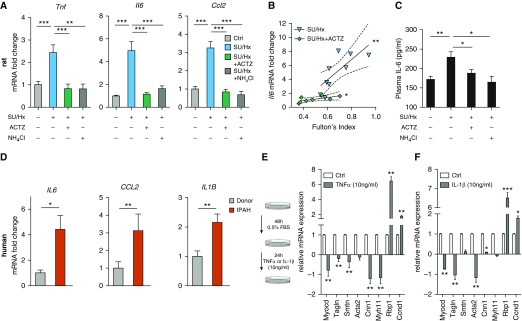
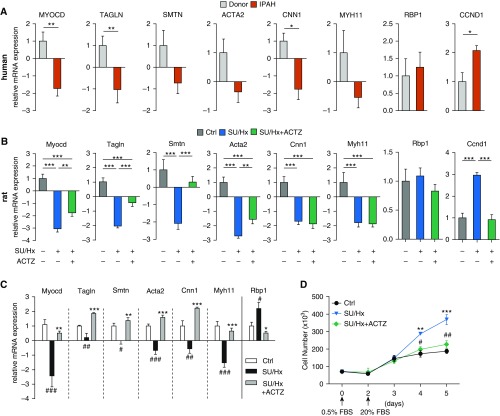
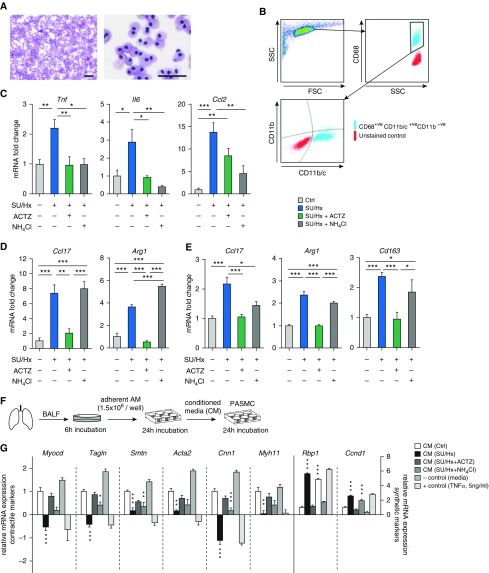
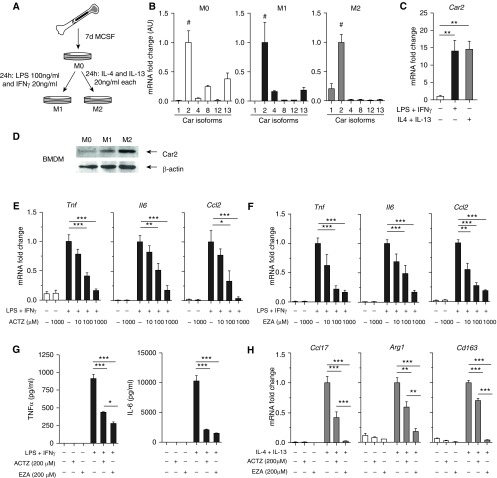
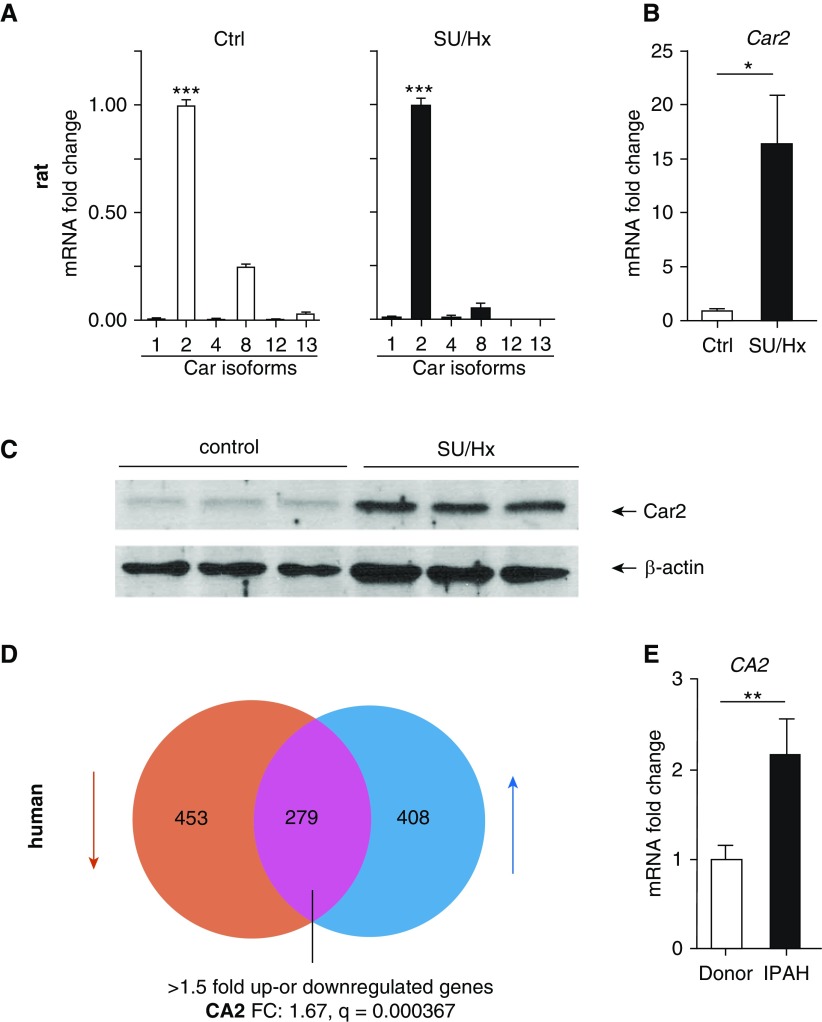
Inflammation and vascular smooth muscle cell (VSMC) phenotypic switching are causally linked to pulmonary arterial hypertension (PAH) pathogenesis. Carbonic anhydrase inhibition induces mild metabolic acidosis and exerts protective effects in hypoxic pulmonary hypertension. Carbonic anhydrases and metabolic acidosis are further known to modulate immune cell activation. To evaluate if carbonic anhydrase inhibition modulates macrophage activation, inflammation, and VSMC phenotypic switching in severe experimental pulmonary hypertension, pulmonary hypertension was assessed in Sugen 5416/hypoxia (SU/Hx) rats after treatment with acetazolamide or ammonium chloride (NH4Cl). We evaluated pulmonary and systemic inflammation and characterized the effect of carbonic anhydrase inhibition and metabolic acidosis in alveolar macrophages and bone marrow-derived macrophages (BMDMs). We further evaluated the treatment effects on VSMC phenotypic switching in pulmonary arteries and pulmonary artery smooth muscle cells (PASMCs) and corroborated some of our findings in lungs and pulmonary arteries of patients with PAH. Both patients with idiopathic PAH and SU/Hx rats had increased expression of lung inflammatory markers and signs of PASMC dedifferentiation in pulmonary arteries. Acetazolamide and NH4Cl ameliorated SU/Hx-induced pulmonary hypertension and blunted pulmonary and systemic inflammation. Expression of carbonic anhydrase isoform 2 was increased in alveolar macrophages from SU/Hx animals, classically (M1) and alternatively (M2) activated BMDMs, and lungs of patients with PAH. Carbonic anhydrase inhibition and acidosis had distinct effects on M1 and M2 markers in BMDMs. Inflammatory cytokines drove PASMC dedifferentiation, and this was inhibited by acetazolamide and acidosis. The protective antiinflammatory effect of acetazolamide in pulmonary hypertension is mediated by a dual mechanism of macrophage carbonic anhydrase inhibition and systemic metabolic acidosis.
Keywords: acetazolamide; acidosis; carbonic anhydrases; lung; macrophages.
Figures
Comment in
-
CA Dreamin': Carbonic Anhydrase Inhibitors, Macrophages, and Pulmonary Hypertension.Am J Respir Cell Mol Biol. 2019 Oct;61(4):412-413. doi: 10.1165/rcmb.2019-0122ED. Am J Respir Cell Mol Biol. 2019. PMID: 30973760 Free PMC article. No abstract available.
References
-
- Farber HW, Loscalzo J. Pulmonary arterial hypertension. N Engl J Med. 2004;351:1655–1665. - PubMed
-
- Savai R, Pullamsetti SS, Kolbe J, Bieniek E, Voswinckel R, Fink L, et al. Immune and inflammatory cell involvement in the pathology of idiopathic pulmonary arterial hypertension. Am J Respir Crit Care Med. 2012;186:897–908. - PubMed
-
- Soon E, Holmes AM, Treacy CM, Doughty NJ, Southgate L, Machado RD, et al. Elevated levels of inflammatory cytokines predict survival in idiopathic and familial pulmonary arterial hypertension. Circulation. 2010;122:920–927. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
