

An unbiased in vitro screen for activating epidermal growth factor receptor mutations
- PMID: 30952700
- PMCID: PMC6579474
- DOI: 10.1074/jbc.RA118.006336
An unbiased in vitro screen for activating epidermal growth factor receptor mutations
Abstract
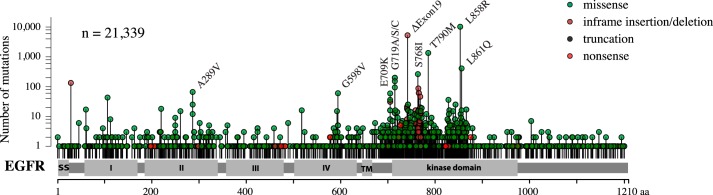
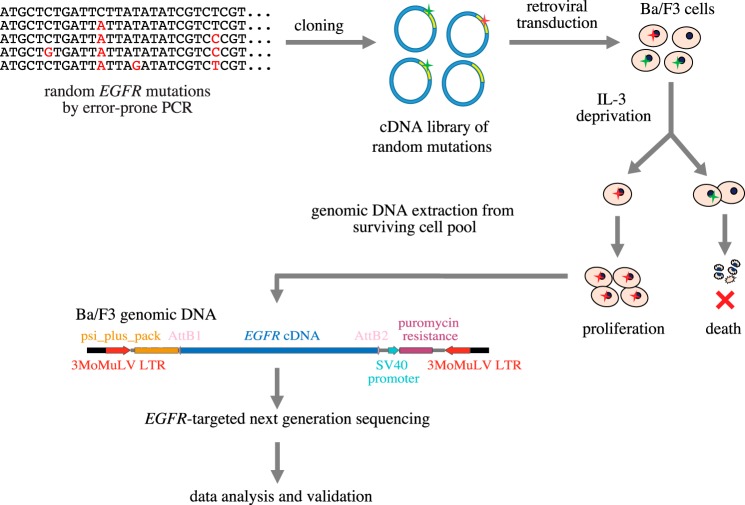
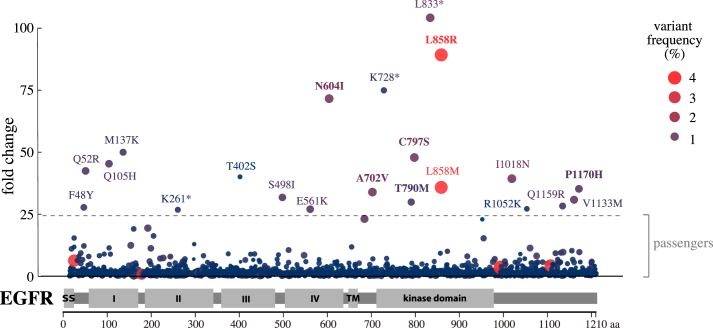
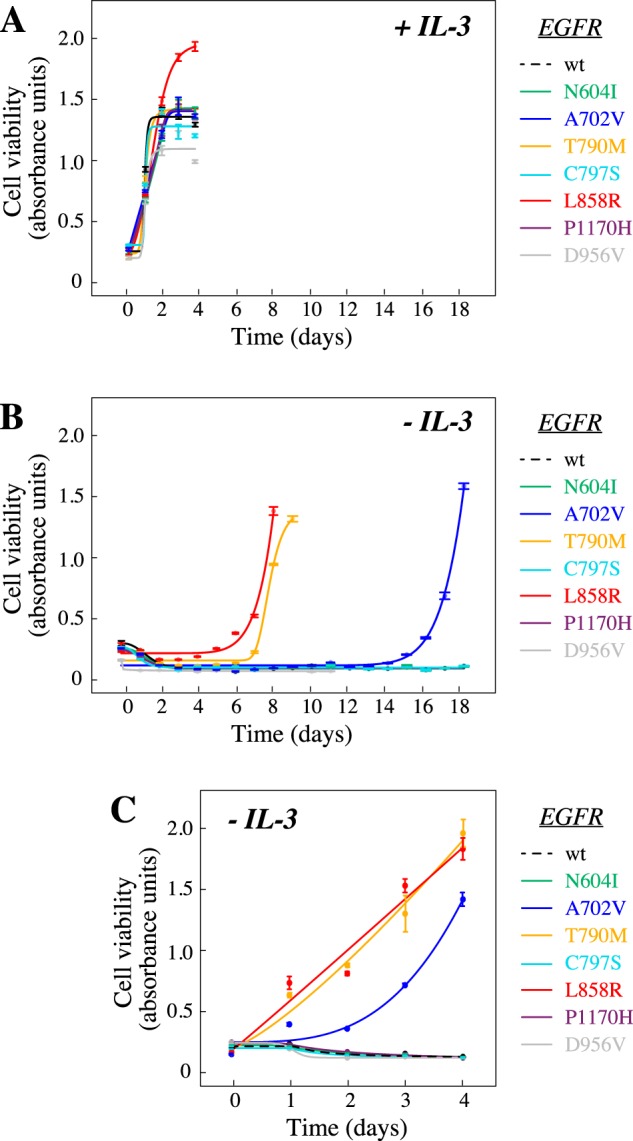
Cancer tissues harbor thousands of mutations, and a given oncogene may be mutated at hundreds of sites, yet only a few of these mutations have been functionally tested. Here, we describe an unbiased platform for the functional characterization of thousands of variants of a single receptor tyrosine kinase (RTK) gene in a single assay. Our
Keywords: ERBB; cancer; cancer biology; clonal selection; directed evolution; driver mutations; epidermal growth factor receptor (EGFR); mutagenesis in vitro; oncogene; passenger mutations; somatic evolution; targeted therapies; tyrosine kinase inhibitors; tyrosine-protein kinase (tyrosine kinase).
© 2019 Chakroborty et al.
Conflict of interest statement
P. A. Jänne has received consulting fees from AstraZeneca, Boehringer Ingelheim, Pfizer, Merrimack Pharmaceuticals, Roche/Genentech, Chugai Pharmaceuticals, ACEA Biosciences, and Ariad Pharmaceuticals and sponsored research funding from Astellas Pharmaceuticals, AstraZeneca, Daiichi Sankyo, and PUMA and receives post-marketing royalties on DFCI-owned intellectual property on EGFR mutations licensed to Lab Corp. K. Elenius has a research agreement with Boehringer Ingelheim and ownership interest in Abomics, Orion, and Roche
Figures
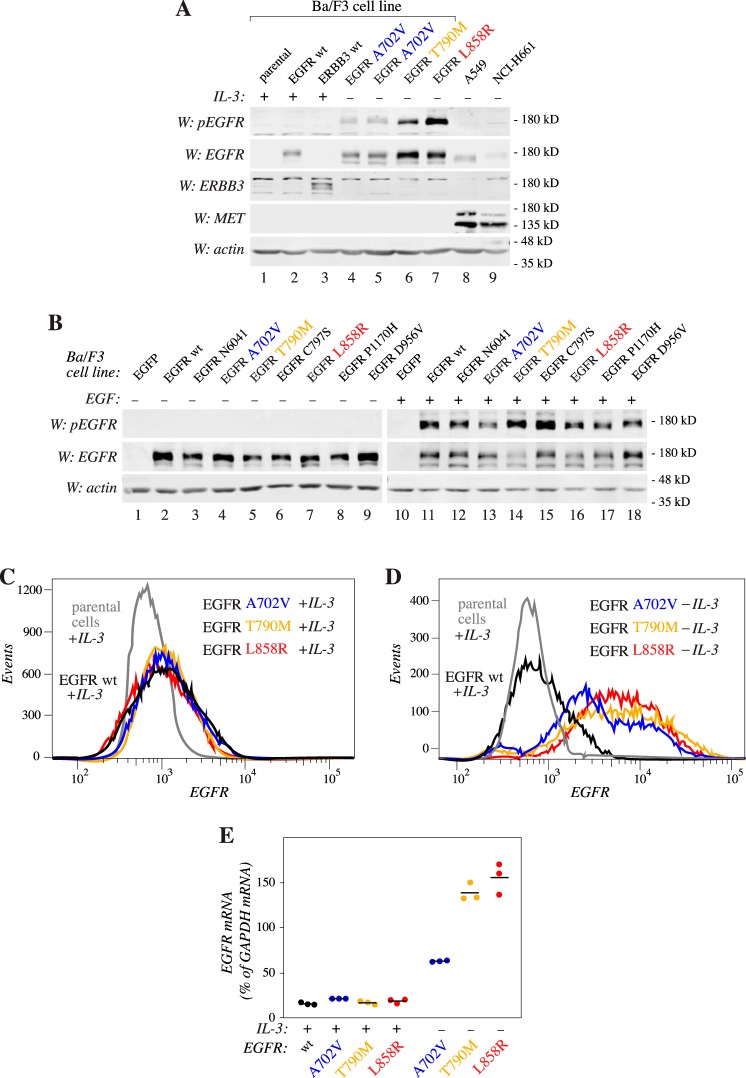
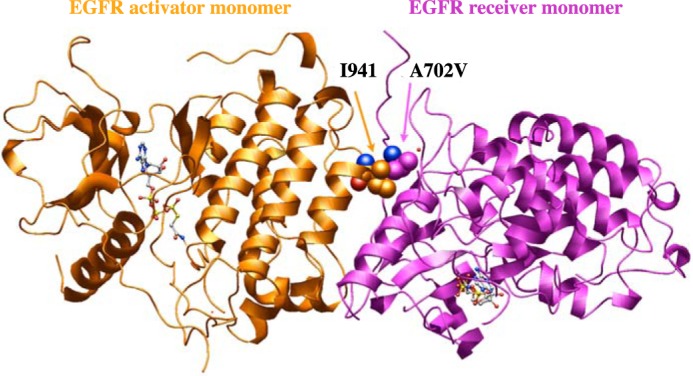
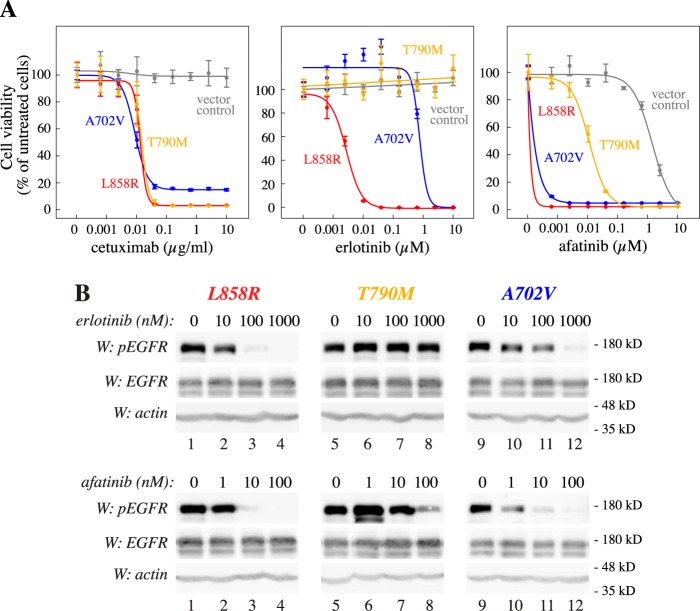
References
-
- Lawrence M. S., Stojanov P., Polak P., Kryukov G. V., Cibulskis K., Sivachenko A., Carter S. L., Stewart C., Mermel C. H., Roberts S. A., Kiezun A., Hammerman P. S., McKenna A., Drier Y., Zou L., et al. (2013) Mutational heterogeneity in cancer and the search for new cancer-associated genes. Nature 499, 214–218 10.1038/nature12213 - DOI - PMC - PubMed
-
- Zehir A., Benayed R., Shah R. H., Syed A., Middha S., Kim H. R., Srinivasan P., Gao J., Chakravarty D., Devlin S. M., Hellmann M. D., Barron D. A., Schram A. M., Hameed M., Dogan S., et al. (2017) Mutational landscape of metastatic cancer revealed from prospective clinical sequencing of 10,000 patients. Nat. Med. 23, 703–713 10.1038/nm.4333 - DOI - PMC - PubMed
-
- Chang M. T., Asthana S., Gao S. P., Lee B. H., Chapman J. S., Kandoth C., Gao J., Socci N. D., Solit D. B., Olshen A. B., Schultz N., and Taylor B. S. (2016) Identifying recurrent mutations in cancer reveals widespread lineage diversity and mutational specificity. Nat. Biotechnol. 34, 155–163 10.1038/nbt.3391 - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
Associated data
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
LinkOut - more resources
Full Text Sources
Medical
Research Materials
Miscellaneous
