

Ancient pathogen genomics as an emerging tool for infectious disease research
- PMID: 30953039
- PMCID: PMC7097038
- DOI: 10.1038/s41576-019-0119-1
Ancient pathogen genomics as an emerging tool for infectious disease research
Abstract
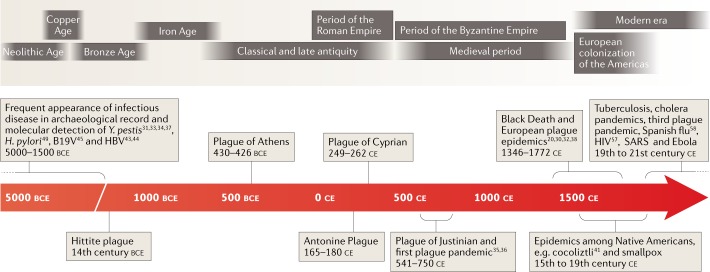
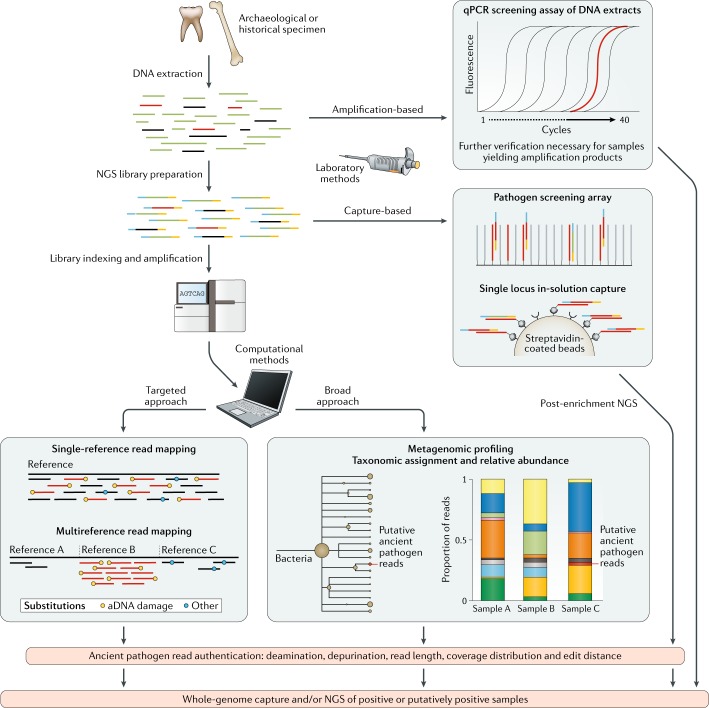
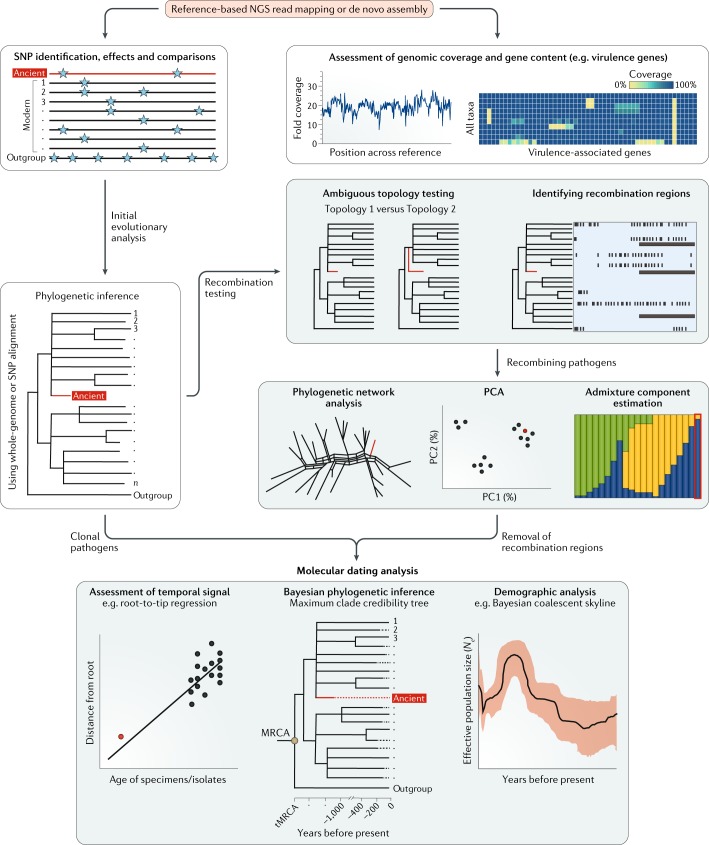
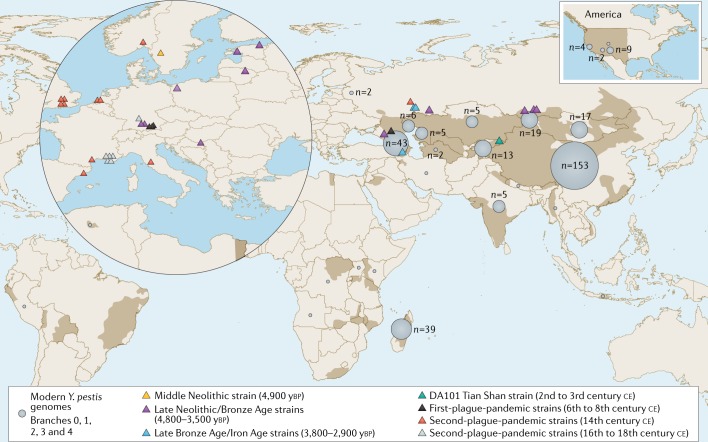
Over the past decade, a genomics revolution, made possible through the development of high-throughput sequencing, has triggered considerable progress in the study of ancient DNA, enabling complete genomes of past organisms to be reconstructed. A newly established branch of this field, ancient pathogen genomics, affords an in-depth view of microbial evolution by providing a molecular fossil record for a number of human-associated pathogens. Recent accomplishments include the confident identification of causative agents from past pandemics, the discovery of microbial lineages that are now extinct, the extrapolation of past emergence events on a chronological scale and the characterization of long-term evolutionary history of microorganisms that remain relevant to public health today. In this Review, we discuss methodological advancements, persistent challenges and novel revelations gained through the study of ancient pathogen genomes.
Conflict of interest statement
The authors declare no competing interests.
Figures
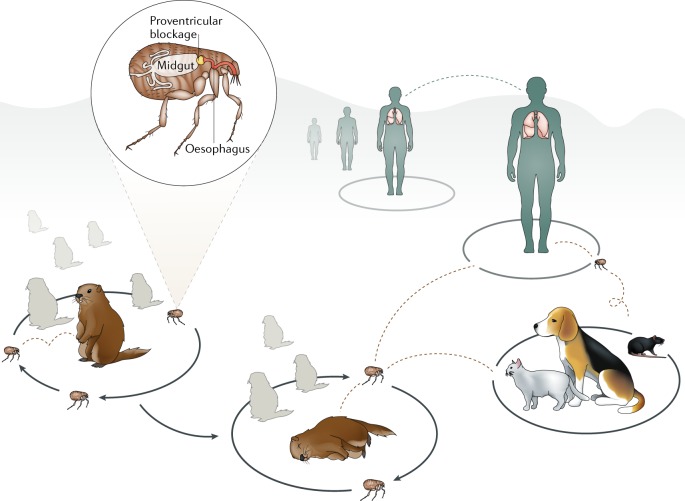
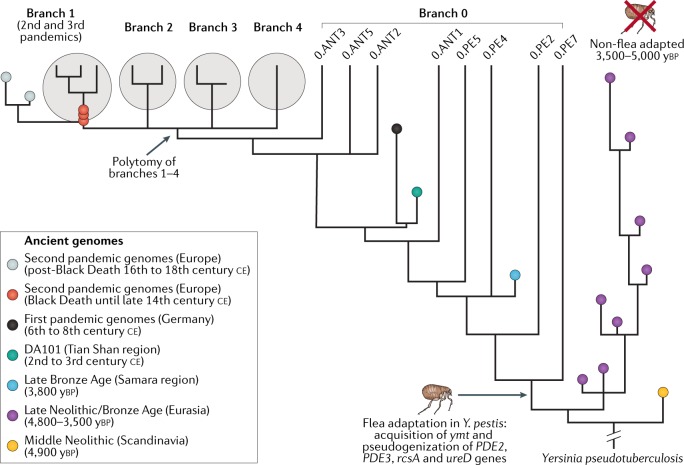
Comment in
-
A genomic approach to microbiology.Nat Rev Genet. 2019 Jun;20(6):311. doi: 10.1038/s41576-019-0131-5. Nat Rev Genet. 2019. PMID: 31101903 No abstract available.
References
-
- Armelagos GJ, Goodman AH, Jacobs KH. The origins of agriculture: population growth during a period of declining health. Popul. Environ. 1991;13:9–22. doi: 10.1007/BF01256568. - DOI
-
- Barrett R, Kuzawa CW, McDade T, Armelagos GJ. Emerging and re-emerging infectious diseases: the third epidemiologic transition. Annu. Rev. Anthropol. 1998;27:247–271. doi: 10.1146/annurev.anthro.27.1.247. - DOI
-
- Ortner, D. J. Identification of Pathological Conditions in Human Skeletal Remains 2nd edn (Academic Press, 2003).
-
- Buikstra, J. E. & Roberts, C. The Global History of Paleopathology: Pioneers and Prospects (Oxford Univ. Press, 2012).
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous
