

Engineering a Model Cell for Rational Tuning of GPCR Signaling
- PMID: 30955892
- PMCID: PMC6476273
- DOI: 10.1016/j.cell.2019.02.023
Engineering a Model Cell for Rational Tuning of GPCR Signaling
Abstract
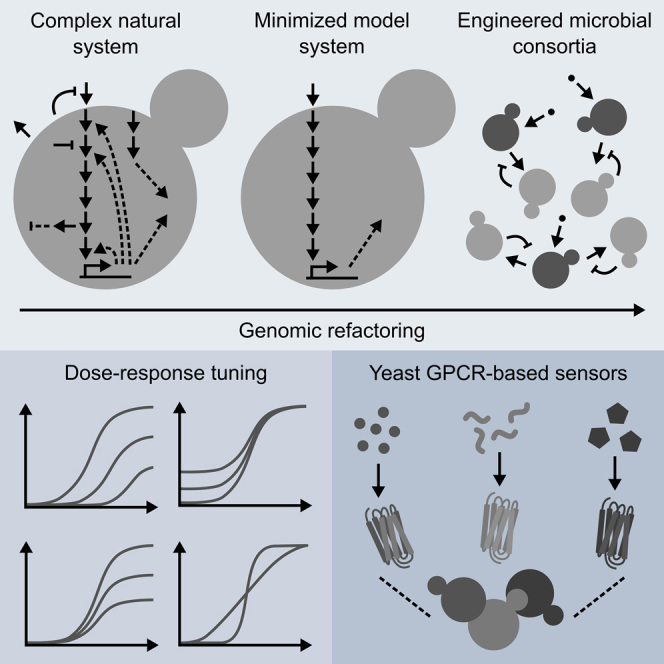
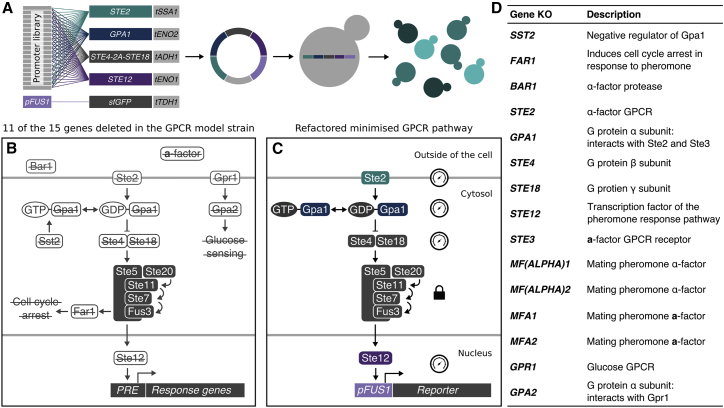
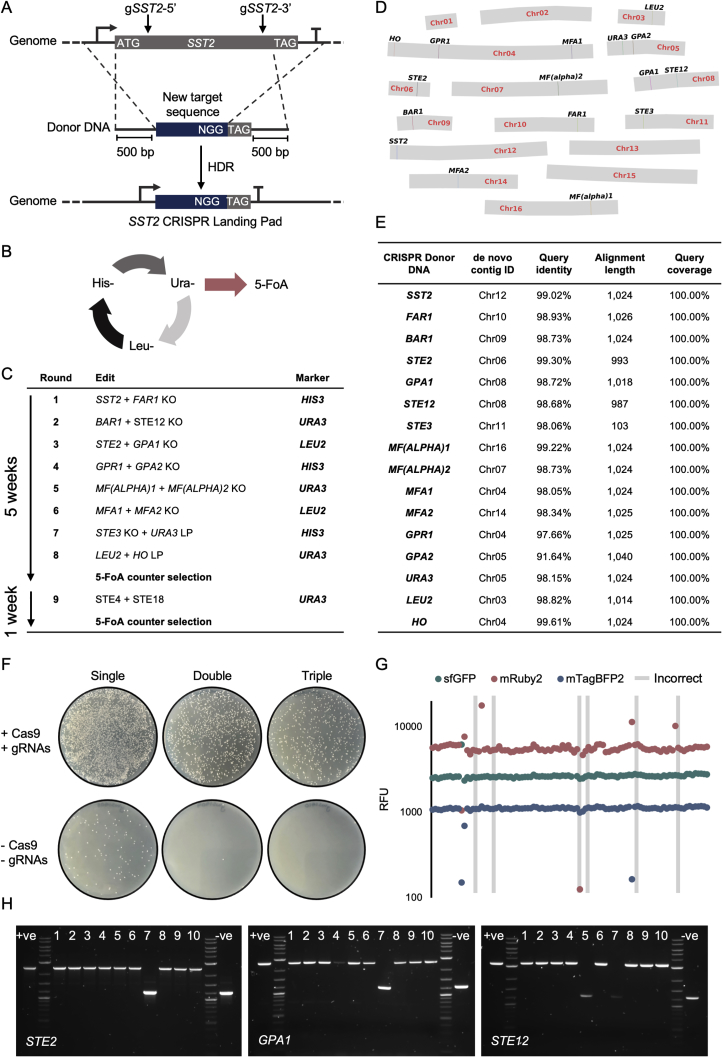
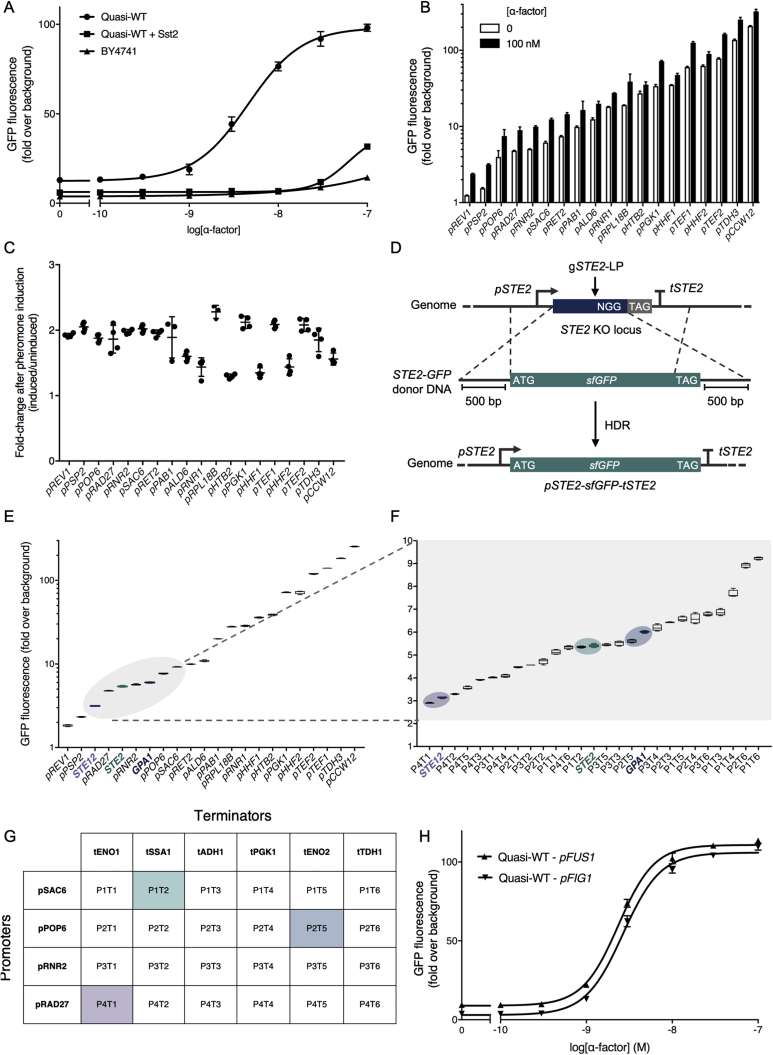
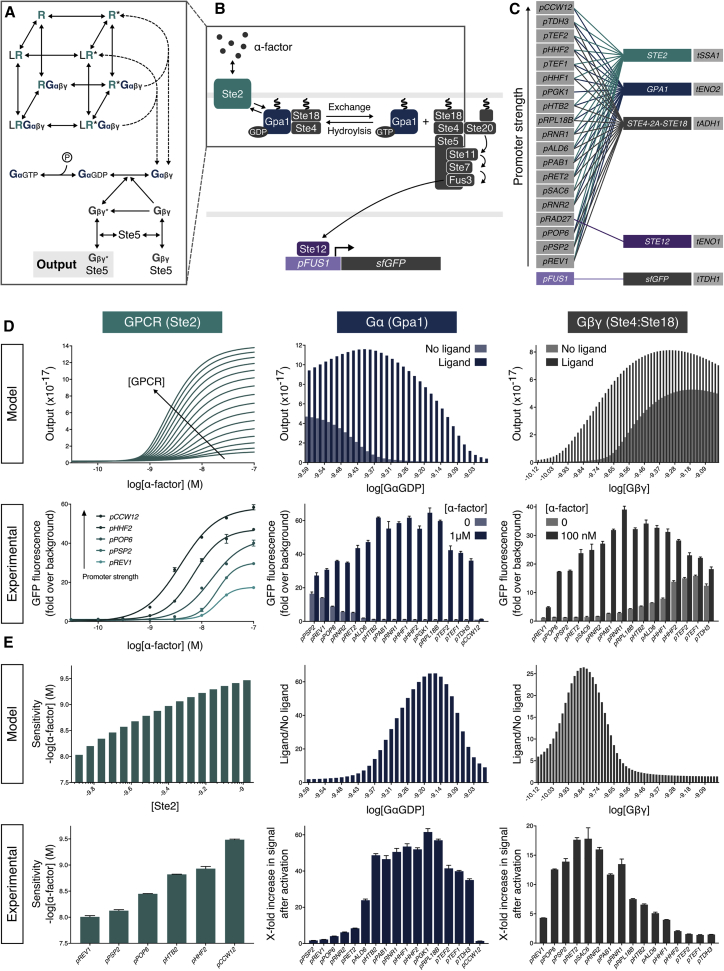
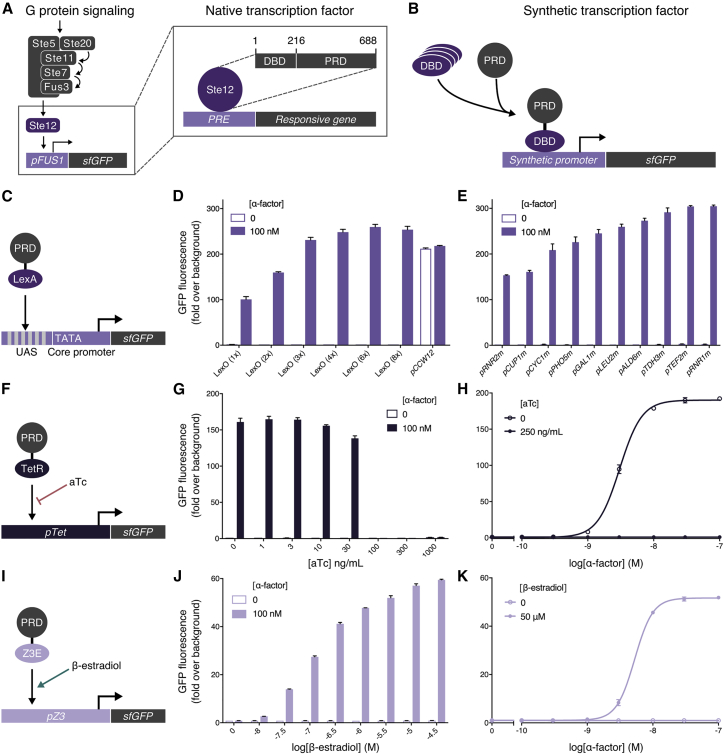
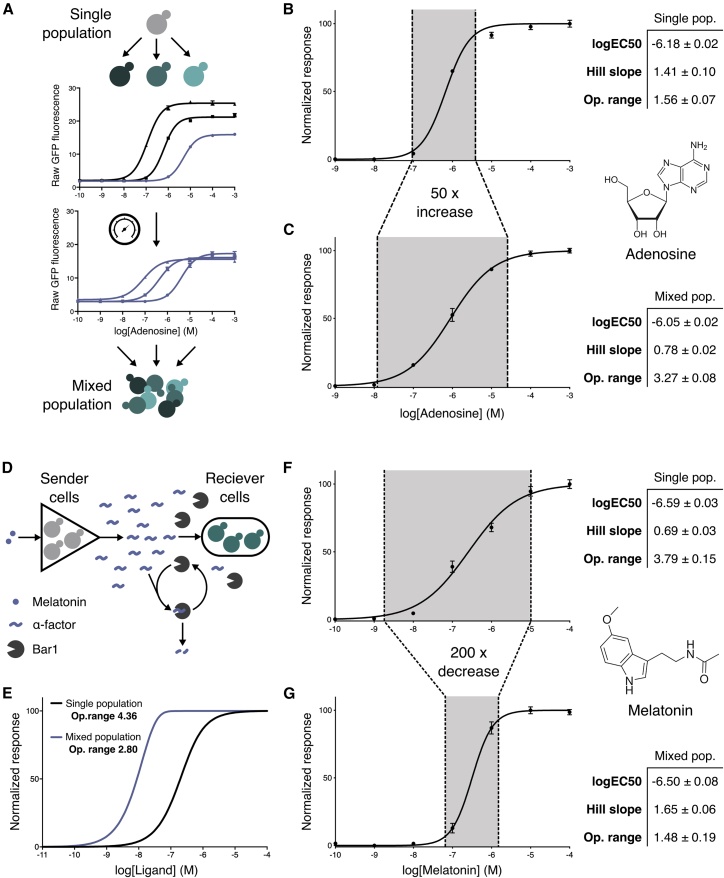
G protein-coupled receptor (GPCR) signaling is the primary method eukaryotes use to respond to specific cues in their environment. However, the relationship between stimulus and response for each GPCR is difficult to predict due to diversity in natural signal transduction architecture and expression. Using genome engineering in yeast, we constructed an insulated, modular GPCR signal transduction system to study how the response to stimuli can be predictably tuned using synthetic tools. We delineated the contributions of a minimal set of key components via computational and experimental refactoring, identifying simple design principles for rationally tuning the dose response. Using five different GPCRs, we demonstrate how this enables cells and consortia to be engineered to respond to desired concentrations of peptides, metabolites, and hormones relevant to human health. This work enables rational tuning of cell sensing while providing a framework to guide reprogramming of GPCR-based signaling in other systems.
Keywords: G protein-coupled receptor; Saccharomyces cerevisiae; biosensor; cell signaling; cell-to-cell communication; genome engineering; synthetic biology.
Copyright © 2019 The Authors. Published by Elsevier Inc. All rights reserved.
Figures







Comment in
-
The Least Mating Pathway: Synthetically Refactoring a Familiar Signaling System for New Applications.Cell. 2019 Apr 18;177(3):521-523. doi: 10.1016/j.cell.2019.04.003. Cell. 2019. PMID: 31002793
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
