

exceRpt: A Comprehensive Analytic Platform for Extracellular RNA Profiling
- PMID: 30956140
- PMCID: PMC7079576
- DOI: 10.1016/j.cels.2019.03.004
exceRpt: A Comprehensive Analytic Platform for Extracellular RNA Profiling
Abstract
Small RNA sequencing has been widely adopted to study the diversity of extracellular RNAs (exRNAs) in biofluids; however, the analysis of exRNA samples can be challenging: they are vulnerable to contamination and artifacts from different isolation techniques, present in lower concentrations than cellular RNA, and occasionally of exogenous origin. To address these challenges, we present exceRpt, the exRNA-processing toolkit of the NIH Extracellular RNA Communication Consortium (ERCC). exceRpt is structured as a cascade of filters and quantifications prioritized based on one's confidence in a given set of annotated RNAs. It generates quality control reports and abundance estimates for RNA biotypes. It is also capable of characterizing mappings to exogenous genomes, which, in turn, can be used to generate phylogenetic trees. exceRpt has been used to uniformly process all ∼3,500 exRNA-seq datasets in the public exRNA Atlas and is available from genboree.org and github.gersteinlab.org/exceRpt.
Keywords: RNA sequencing; RNA-seq; bioinformatics; bioinformatics tool; exRNAs; extracellular RNA; genomics; pipeline; transcriptome.
Copyright © 2019 The Authors. Published by Elsevier Inc. All rights reserved.
Figures
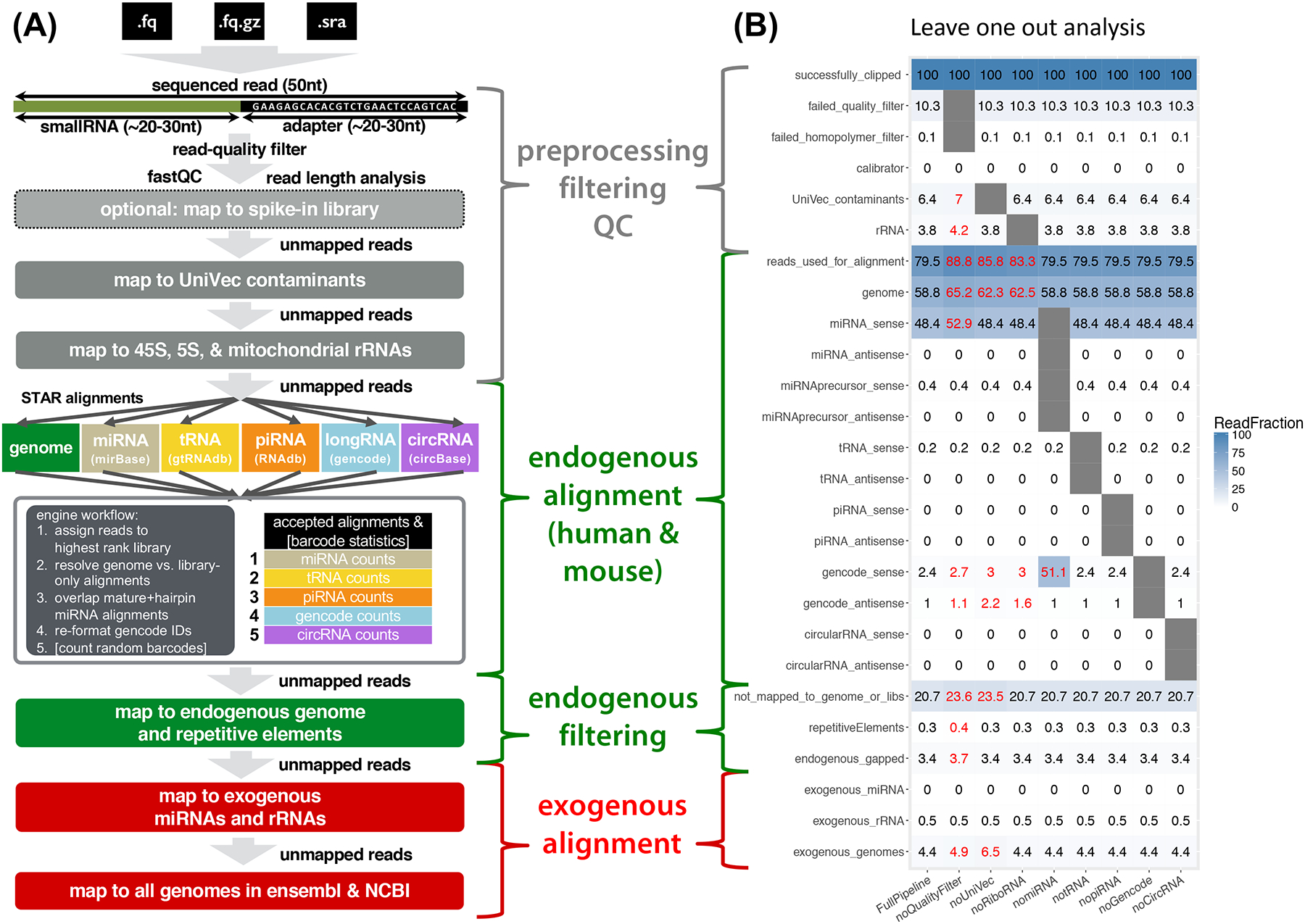
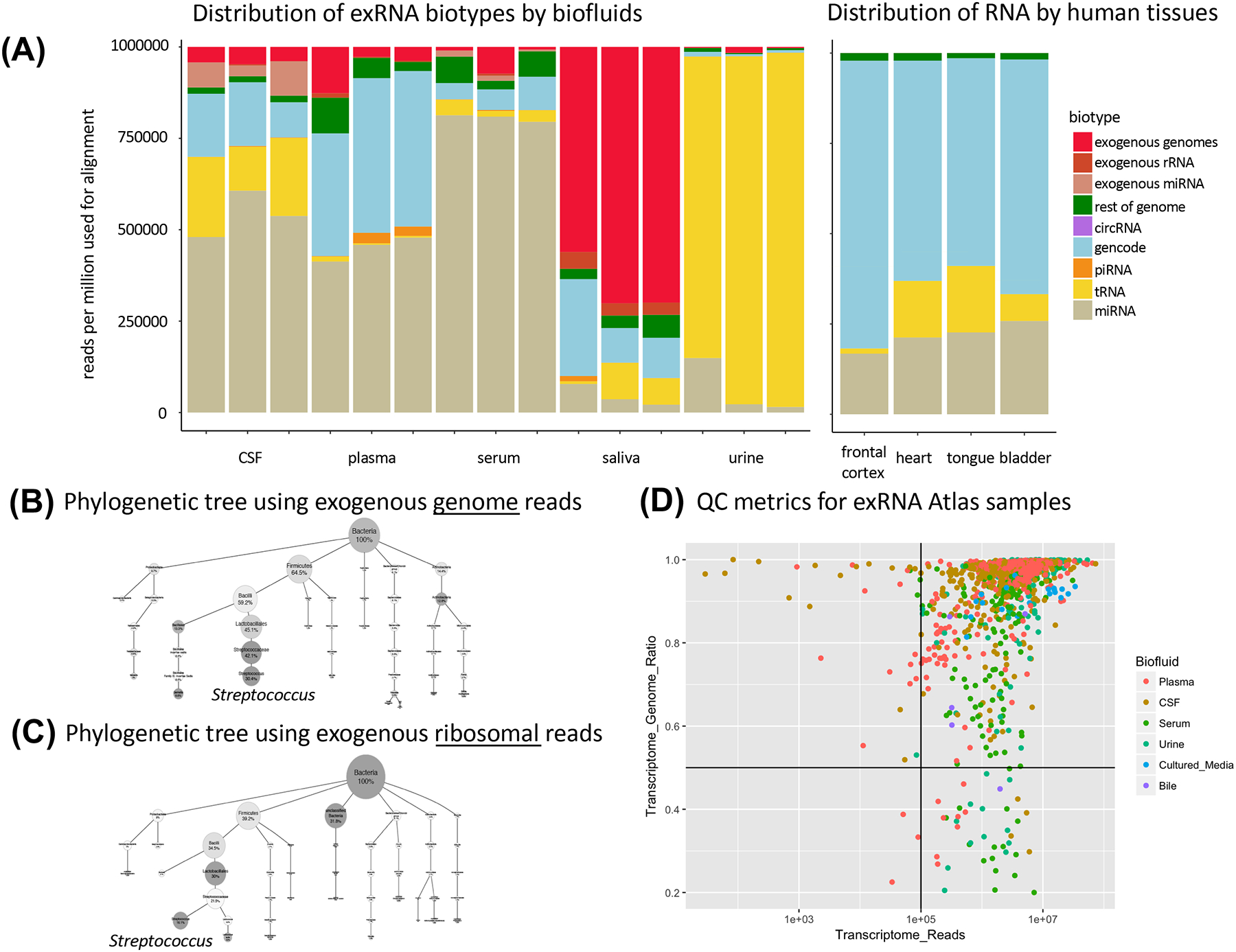
Comment in
-
Mapping Extracellular RNA Sheds Lights on Distinct Carriers.Cell. 2019 Apr 4;177(2):228-230. doi: 10.1016/j.cell.2019.03.027. Cell. 2019. PMID: 30951666
References
-
- Akat KM, Moore-McGriff D, Morozov P, Brown M, Gogakos T, Correa Da Rosa J, Mihailovic A, Sauer M, Ji R, Ramarathnam A, et al. (2014). Comparative RNA-sequencing analysis of myocardial and circulating small RNAs in human heart failure and their utility as biomarkers. Proc Natl Acad Sci U S A 111, 11151–11156. - PMC - PubMed
-
- Barturen G, Rueda A, Hamberg M, Alganza A, Lebron R, Kotsyfakis M, et al. (2014). sRNAbench: profiling of small RNAs and its sequence variants in single or multi-species high-throughput experiments. Methods in Next Generation Sequencing: Methods in Next Generation Sequencing; 2014.
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
