

Molecular diagnosis of glycogen storage disease type I: a review
- PMID: 30956637
- PMCID: PMC6449677
Molecular diagnosis of glycogen storage disease type I: a review
Abstract
Glycogen storage disease type I (GSD I) is a relatively rare metabolic disease with variable clinical intensity. It is caused by deficient activity of the glucose 6-phosphatase enzyme (GSD Ia) or a deficiency in the microsomal transport proteins for glucose 6-phosphate (GSD Ib). We searched the most recent English literature (1997-2017) regarding any article with the key word of "glycogen storage disease type I" in PubMed, Science Direct, Scopus, EMBASE, and Google Scholar. We will present all of the published articles about the molecular genetic characteristics and old-to-new diagnostic methods used to identify GSD I in regard of methodology, advantages and disadvantages. Diagnosis of GSD type I and its variants is challenging because it is a genetically heterogeneous disorder. Many molecular methods have been used to diagnose GSD I most of which have been based on mutation detection. Therefore, we discuss complete aspects of all of the molecular diagnostic tests, which have been used in GSD type I so far. With the advent of high throughput advanced molecular tests, molecular diagnosis is going to be an important platform for the diagnosis of storage and metabolic diseases such as GSD type I. Next-generation sequencing, in combination with the biochemical tests and clinical signs and symptoms create an accurate, reliable and feasible method. It can overcome the difficulties by the diagnosis of diseases with broad clinical and genetic heterogeneity.
Keywords: DNA mutational analysis; glucose-6-phosphatase-alpha; glucose-6-phosphate translocase; glycogen storage disease type I; molecular genetics diagnosis.
Figures

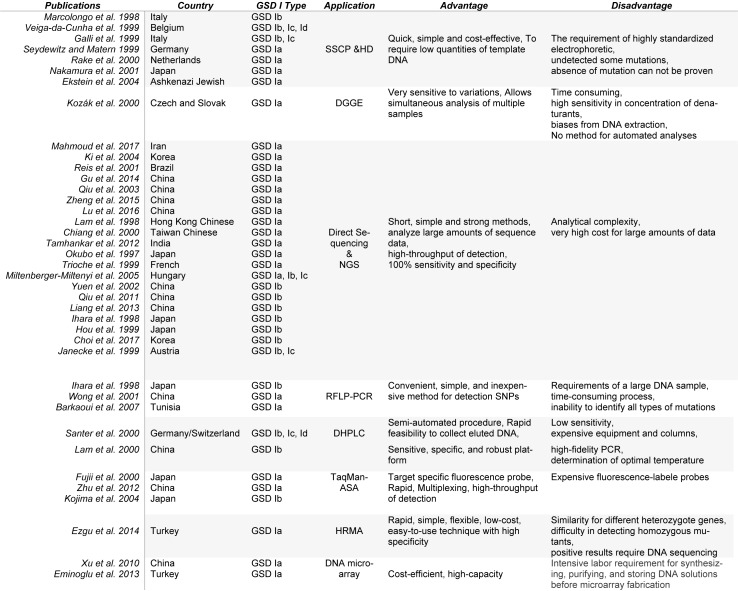
References
-
- Bagheri Lankarani K, Eshraghian K, Malek-Hosseini SA, Janghorban P, Geramizadeh B, Eshraghian A. Outcomes of liver transplantation for patients with acute liver failure. Arch Iran Med. 2013;16:64–68. - PubMed
-
- Barkaoui E, Cherif W, Tebib N, Charfeddine C, Ben Rhouma F, Azzouz H, et al. Mutation spectrum of glycogen storage disease type Ia in Tunisia: implication for molecular diagnosis. J Inherit Metab Dis. 2007;30:989–993. - PubMed
-
- Børresen AL. Mismatch detection using heteroduplex analysis. Curr Protoc Hum Genet. 2002;Chapter 7:Unit 7.3. - PubMed
-
- Chen YT. The glycogen storage diseases. In: Scriver CR, Beaudet AL, Sly WS, Valle D, Childs B, Kinzler KW, et al., editors. The metabolic and molecular bases of inherited disease. New York: McGraw-Hill; 2003. pp. 1521–1551.
-
- Chiang SC, Lee YM, Chang MH, Wang TR, Ko TM, Hwu WL. Glucose-6-phosphatase gene mutations in Taiwan Chinese patients with glycogen storage disease type Ia. J Hum Genet. 2000;45:197–199. - PubMed
Publication types
LinkOut - more resources
Full Text Sources