

Modulation of mTOR and CREB pathways following mGluR5 blockade contribute to improved Huntington's pathology in zQ175 mice
- PMID: 30961637
- PMCID: PMC6454676
- DOI: 10.1186/s13041-019-0456-1
Modulation of mTOR and CREB pathways following mGluR5 blockade contribute to improved Huntington's pathology in zQ175 mice
Abstract
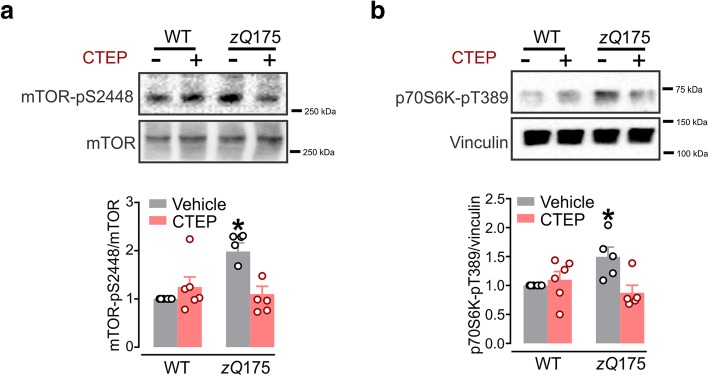
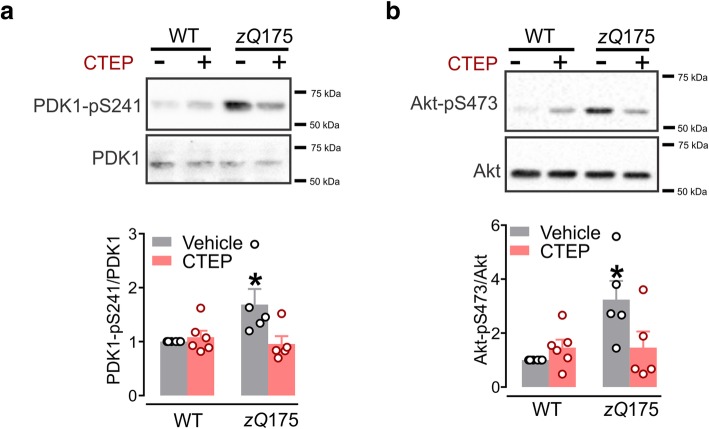
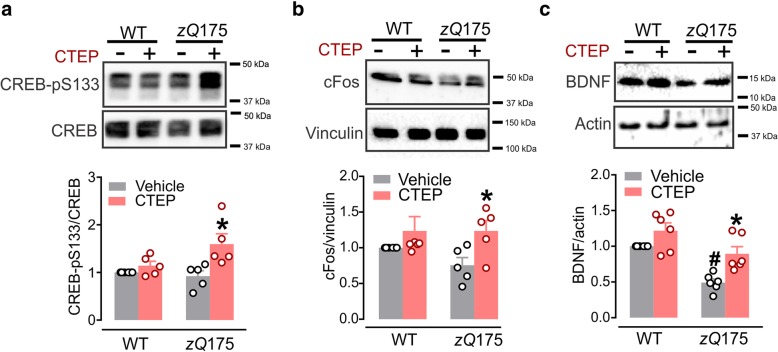
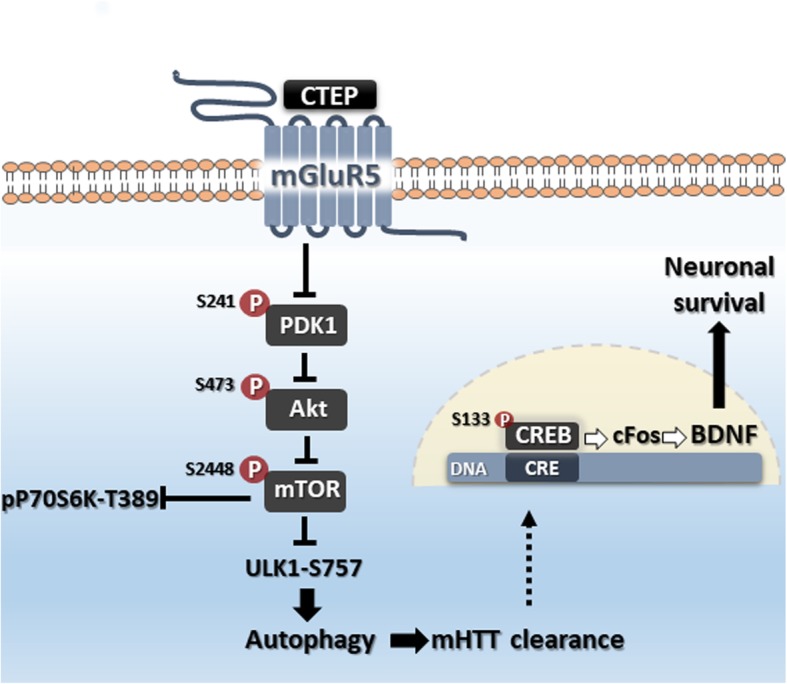
Huntington's disease (HD) is a neurodegenerative disorder caused by a genetic abnormality in the huntingtin gene that leads to a polyglutamine repeat expansion of the huntingtin protein. The cleaved polyglutamine expansion of mutant huntingtin (mHTT) protein can form aggregates strongly correlated with HD progression. We have previously shown that the inhibition of mGluR5 using CTEP, a selective negative allosteric mGluR5 modulator, can delay disease progression and reduce in mHTT aggregates in the zQ175 mouse model of HD. This was paralleled by enhanced catalytic activity of Unc-51-like kinase 1 (ULK1), a kinase modulated by mammalian target of rapamycin (mTOR) and key regulator of autophagy initiation. In the present study, we show that CTEP can correct aberrant phosphoinositide 3-kinase (PI3K)/Akt/mTOR signaling detected in zQ175 mice that may underlie the enhanced ULK1 activity and activation of autophagy. We also show that CTEP can facilitate cAMP response element-binding protein (CREB)-mediated expression of brain-derived neurotrophic factor (BDNF) to foster neuronal survival and reduce apoptosis. Taken together, our findings provide the molecular evidence for how targeting mGluR5 using a well-tolerated selective NAM can mitigate two critical mechanisms of neurodegeneration, autophagy and apoptosis.
Keywords: BDNF; CTEP; Huntington’s disease; ULK1; autophagy; mGluR5; mHTT; mTOR; zQ175.
Conflict of interest statement
Ethics approval
All animal experiments were conducted in accordance with University of Ottawa animal care committees.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Miscellaneous
