

Shallow Sparsely-Connected Autoencoders for Gene Set Projection
- PMID: 30963076
- PMCID: PMC6417803
Shallow Sparsely-Connected Autoencoders for Gene Set Projection
Abstract
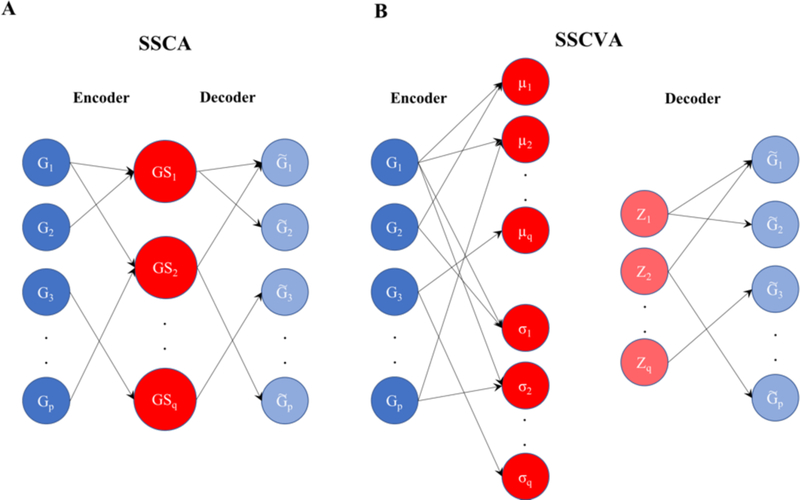
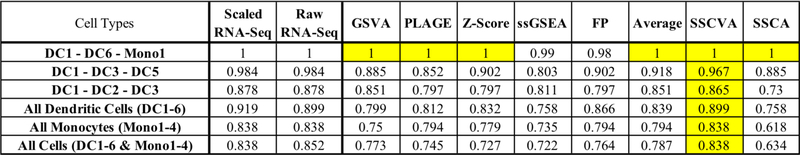
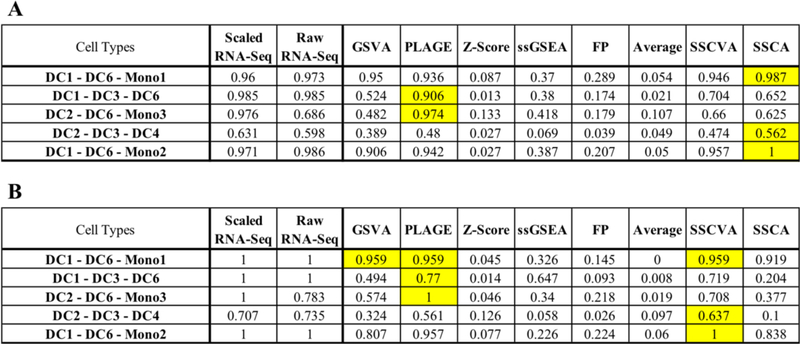
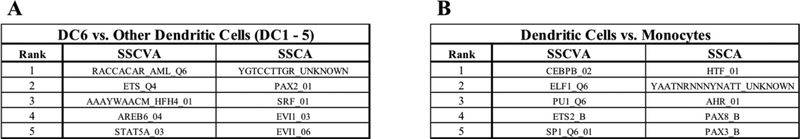
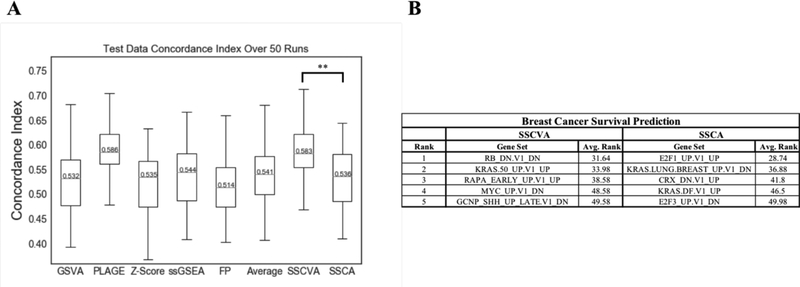
When analyzing biological data, it can be helpful to consider gene sets, or predefined groups of biologically related genes. Methods exist for identifying gene sets that are differential between conditions, but large public datasets from consortium projects and single-cell RNA-Sequencing have opened the door for gene set analysis using more sophisticated machine learning techniques, such as autoencoders and variational autoencoders. We present shallow sparsely-connected autoencoders (SSCAs) and variational autoencoders (SSCVAs) as tools for projecting gene-level data onto gene sets. We tested these approaches on single-cell RNA-Sequencing data from blood cells and on RNA-Sequencing data from breast cancer patients. Both SSCA and SSCVA can recover known biological features from these datasets and the SSCVA method often outperforms SSCA (and six existing gene set scoring algorithms) on classification and prediction tasks.
Keywords: autoencoder; gene set; single-cell RNA-Sequencing; variational autoencoder.
Figures
References
-
- Weinstein JN, Collisson E. a, Mills GB, Shaw KRM, Ozenberger B. a, Ellrott K, Shmulevich I, Sander C & Stuart JM. Nat. Genet 45, 1113 (2013).
-
- Tang F, Barbacioru C, Wang Y, Nordman E, Lee C, Xu N, Wang X, Bodeau J, Tuch BB, Siddiqui A, Lao K & Surani MA Nat. Methods 6, 377 (2009). - PubMed
-
- Liou CY, Huang JC & Yang WC in Neurocomputing 71, 3150 (2008).
-
- Kingma DP & Welling M Ppt (2013). doi: 10.1051/0004-6361/201527329 - DOI
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources