

An update on the central nervous system manifestations of neurofibromatosis type 1
- PMID: 30963251
- PMCID: PMC6819239
- DOI: 10.1007/s00401-019-02002-2
An update on the central nervous system manifestations of neurofibromatosis type 1
Abstract
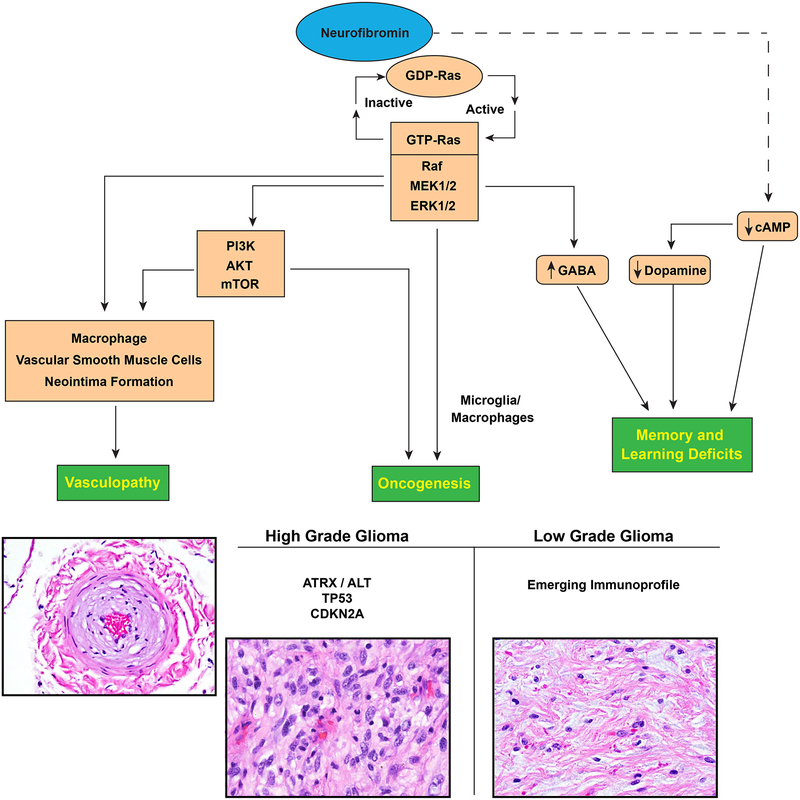
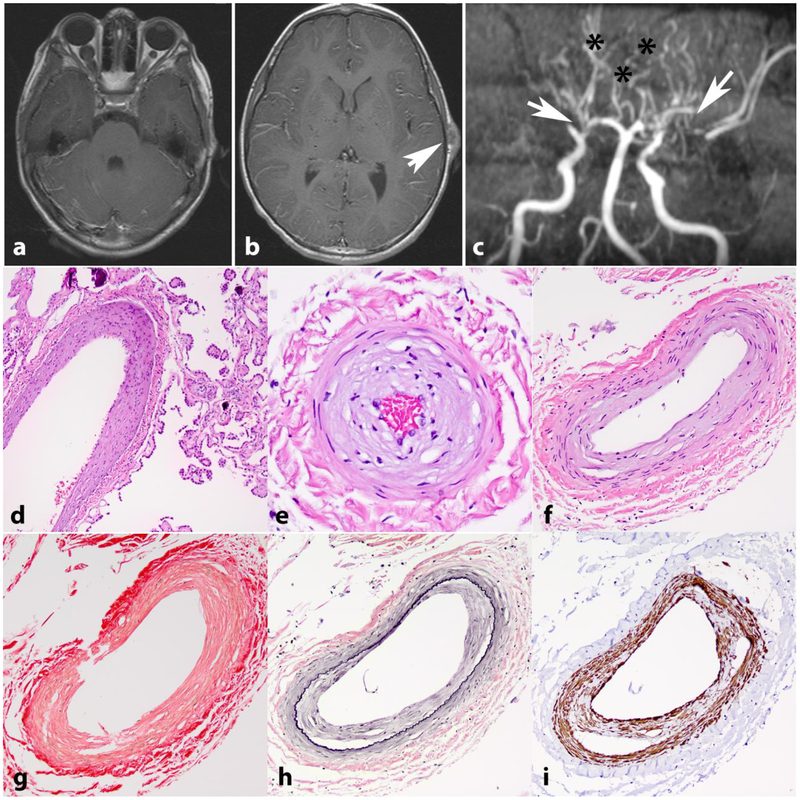
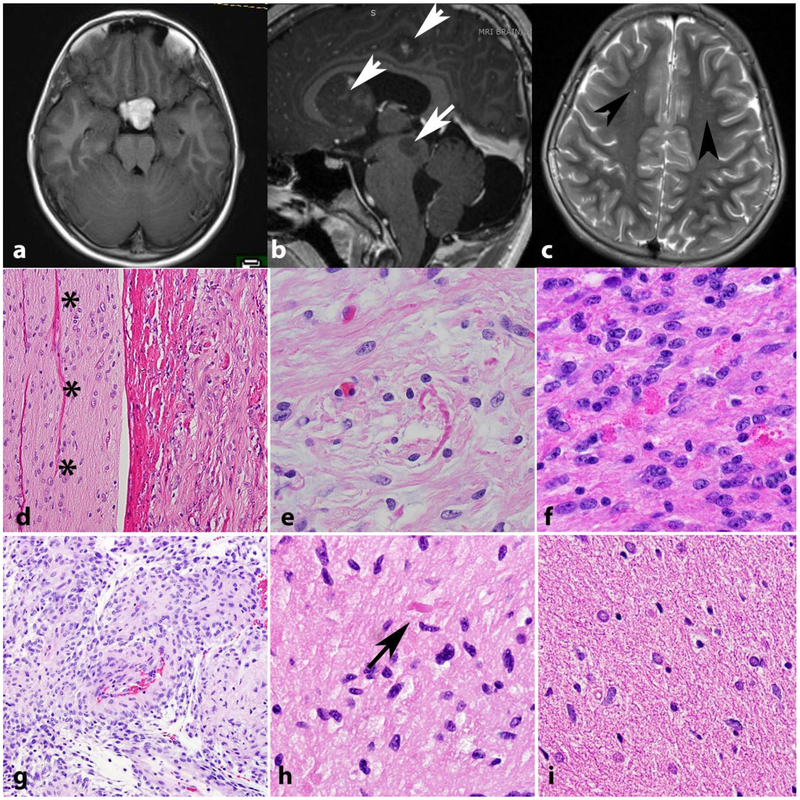
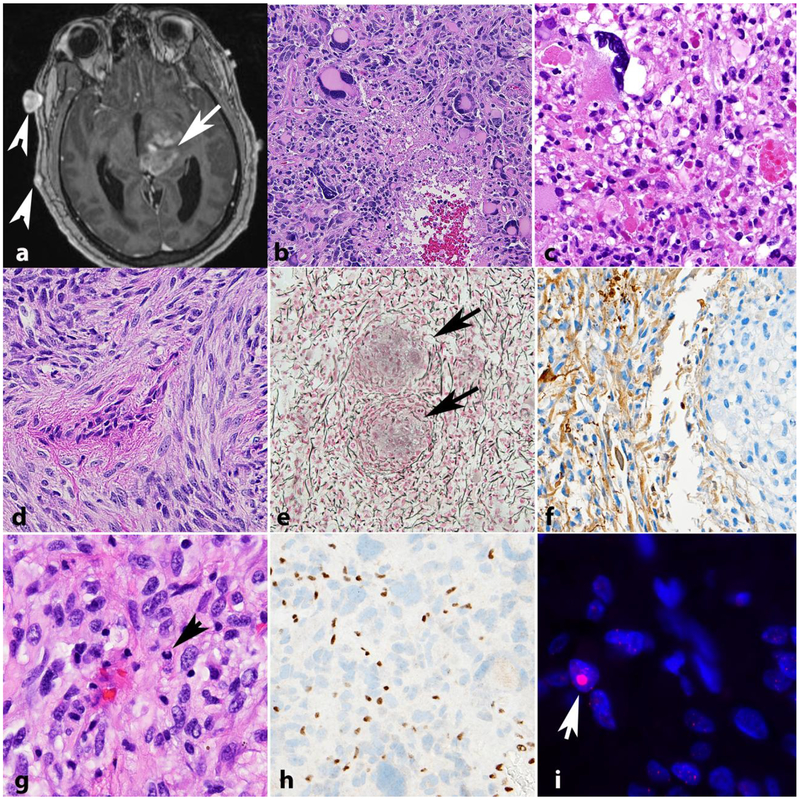
Neurofibromatosis 1 (NF1) is an autosomal dominant genetic disorder that presents with variable phenotypes as a result of mutations in the neurofibromatosis type 1 (NF1) gene and subsequently, abnormal function of the protein product, neurofibromin. Patients with NF1 are at increased risk for central nervous system (CNS) manifestations including structural, functional, and neoplastic disease. The mechanisms underlying the varied manifestations of NF1 are incompletely understood, but the loss of functional neurofibromin, resulting in sustained activation of the oncoprotein RAS, is responsible for tumorigenesis throughout the body, including the CNS. Much of our understanding of NF1-related CNS manifestations is from a combination of data from animal models and natural history studies of people with NF1 and CNS disease. Data from animal models suggest the importance of both Nf1 mutations and somatic genetic alterations, such as Tp53 loss, for development of neoplasms, as well as the role of the timing of the acquisition of such alterations on the variability of CNS manifestations. A variety of non-neoplastic structural (macrocephaly, hydrocephalus, aqueductal stenosis, and vasculopathy) and functional (epilepsy, impaired cognition, attention deficits, and autism spectrum disorder) abnormalities occur with variable frequency in individuals with NF1. In addition, there is increasing evidence that similar appearing CNS neoplasms in people with and without the NF1 syndrome are due to distinct oncogenic pathways. Gliomas in people with NF1 show alterations in the RAS/MAPK pathway, generally in the absence of BRAF alterations (common to sporadic pilocytic astrocytomas) or IDH or histone H3 mutations (common to diffuse gliomas subsets). A subset of low-grade astrocytomas in these patients remain difficult to classify using standard criteria, and occasionally demonstrate morphologic features resembling subependymal giant cell astrocytomas that afflict patients with tuberous sclerosis complex ("SEGA-like astrocytomas"). There is also emerging evidence that NF1-associated high-grade astrocytomas have frequent co-existing alterations such as ATRX mutations and an alternative lengthening of telomeres (ALT) phenotype responsible for unique biologic properties. Ongoing efforts are seeking to improve diagnostic accuracy for CNS neoplasms in the setting of NF1 versus sporadic tumors. In addition, MEK inhibitors, which act on the RAS/MAPK pathway, continue to be studied as rational targets for the treatment of NF1-associated tumors, including CNS tumors.
Keywords: Brain tumor; Glioma; Hydrocephalus; Neurofibromatosis; Neurofibromin; Seizure; Vasculopathy.
Figures
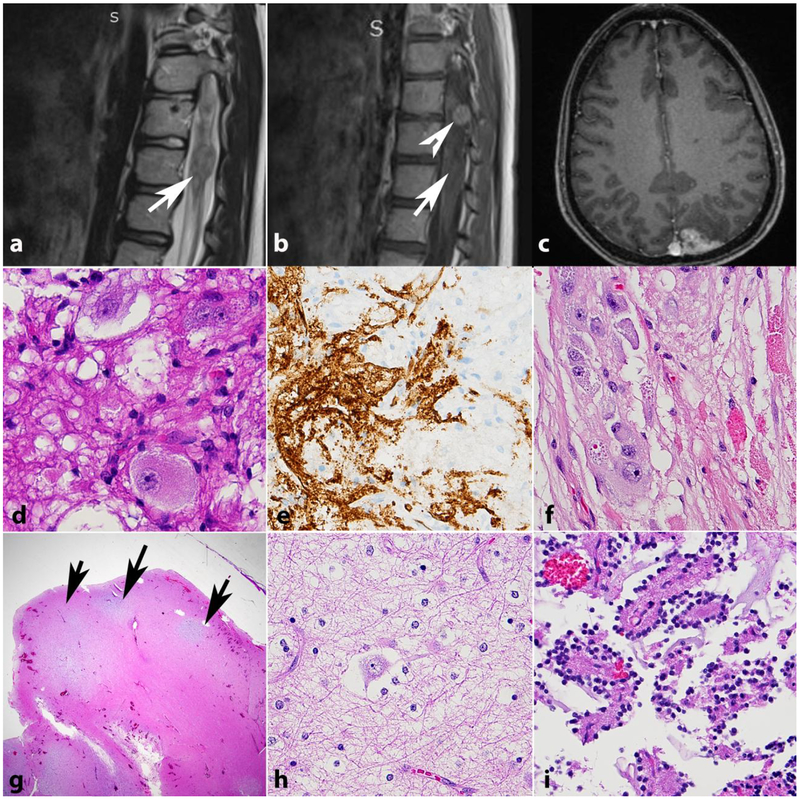
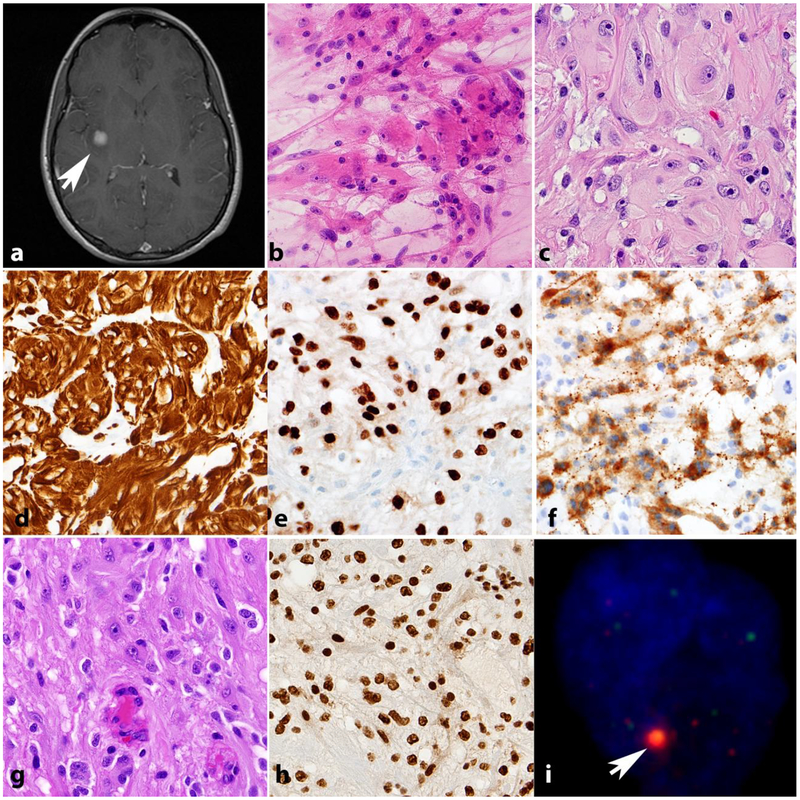
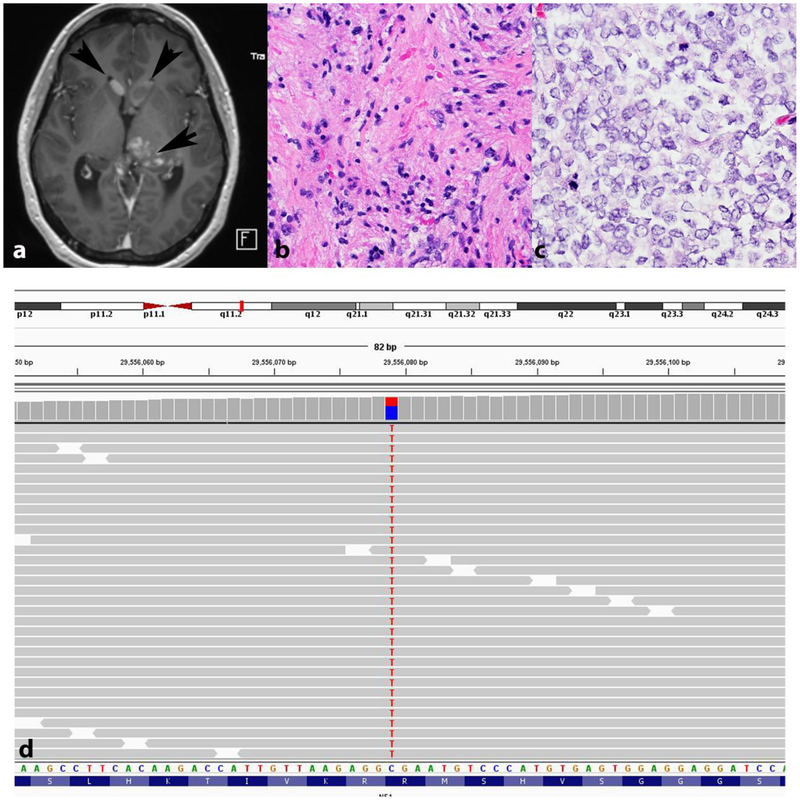
References
-
- Avery RA, Hwang EI, Ishikawa H, Acosta MT, Hutcheson KA, Santos D, Zand DJ, Kilburn LB, Rosenbaum KN, Rood BR, Schuman JS, Packer RJ (2014) Handheld optical coherence tomography during sedation in young children with optic pathway gliomas. JAMA Ophthalmol 132:265–271. doi:10.1001/jamaophthalmol.2013.7649 - DOI - PMC - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
