

Increased MDR1 Transporter Expression in Human Brain Endothelial Cells Through Enhanced Histone Acetylation and Activation of Aryl Hydrocarbon Receptor Signaling
- PMID: 30963442
- PMCID: PMC6728213
- DOI: 10.1007/s12035-019-1565-7
Increased MDR1 Transporter Expression in Human Brain Endothelial Cells Through Enhanced Histone Acetylation and Activation of Aryl Hydrocarbon Receptor Signaling
Abstract
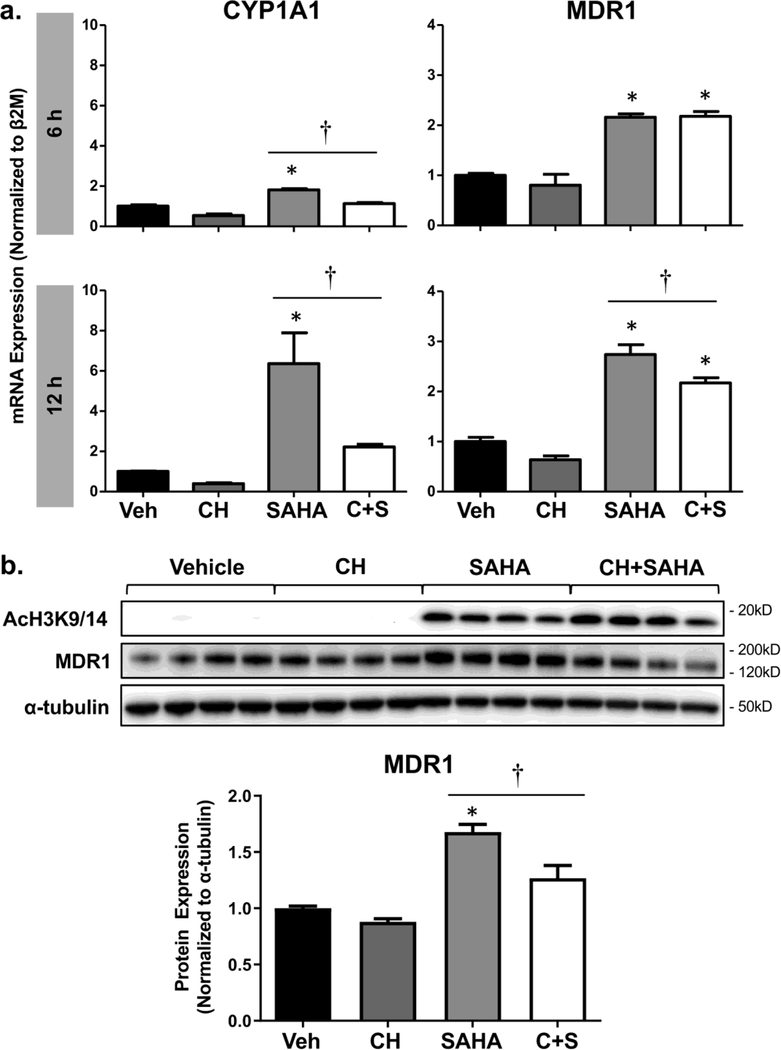
Multidrug resistance protein 1 (MDR1, ABCB1, P-glycoprotein) is a critical efflux transporter that extrudes chemicals from the blood-brain barrier (BBB) and limits neuronal exposure to xenobiotics. Prior studies in malignant cells demonstrated that MDR1 expression can be altered by inhibition of histone deacetylases (HDAC), enzymes that modify histone structure and influence transcription factor binding to DNA. Here, we sought to identify the mechanisms responsible for the up-regulation of MDR1 by HDAC inhibitors in human BBB cells. Immortalized human brain capillary endothelial (hCMEC/D3) cells were treated with HDAC inhibitors and assessed for MDR1 expression and function. Of the HDAC inhibitors profiled, valproic acid (VPA), apicidin, and suberoylanilide hydroxamic acid (SAHA) increased MDR1 mRNA and protein levels by 30-200%, which corresponded with reduced intracellular accumulation of the MDR1 substrate rhodamine 123. Interestingly, induction of MDR1 mRNA by HDAC inhibitors mirrored increases in the expression of the aryl hydrocarbon receptor (AHR) and its target gene cytochrome P450 1A1. To explore the role of AHR in HDAC inhibitor-mediated regulation of MDR1, a pharmacological activator (β-naphthoflavone, βNF) and inhibitor (CH-223191, CH) of AHR were tested. The induction of MDR1 in cells treated with SAHA was amplified by βNF and attenuated by CH. Furthermore, SAHA increased the binding of acetylated histone H3K9/K14 and AHR proteins to regions of the MDR1 promoter that contain AHR response elements. In conclusion, HDAC inhibitors up-regulate the expression and activity of the MDR1 transporter in human brain endothelial cells by increasing histone acetylation and facilitating AHR binding at the MDR1 promoter.
Keywords: Aryl hydrocarbon receptor; Blood–brain barrier; HDAC; MDR1; Transport.
Conflict of interest statement
Figures







Similar articles
-
Epigenetic Regulation of Multidrug Resistance Protein 1 and Breast Cancer Resistance Protein Transporters by Histone Deacetylase Inhibition.Drug Metab Dispos. 2020 Jun;48(6):459-480. doi: 10.1124/dmd.119.089953. Epub 2020 Mar 19. Drug Metab Dispos. 2020. PMID: 32193359 Free PMC article. Review.
-
Brain region-specific regulation of histone acetylation and efflux transporters in mice.J Biochem Mol Toxicol. 2019 Jun;33(6):e22318. doi: 10.1002/jbt.22318. Epub 2019 Mar 21. J Biochem Mol Toxicol. 2019. PMID: 30897286 Free PMC article.
-
HDAC6 modulates Hsp90 chaperone activity and regulates activation of aryl hydrocarbon receptor signaling.J Biol Chem. 2009 Mar 20;284(12):7436-45. doi: 10.1074/jbc.M808999200. Epub 2009 Jan 21. J Biol Chem. 2009. Retraction in: J Biol Chem. 2020 Jan 3;295(1):297. doi: 10.1074/jbc.W119.012142. PMID: 19158084 Free PMC article. Retracted.
-
Aza-PBHA, a potent histone deacetylase inhibitor, inhibits human gastric-cancer cell migration via PKCα-mediated AHR-HDAC interactions.Biochim Biophys Acta Mol Cell Res. 2020 Feb;1867(2):118564. doi: 10.1016/j.bbamcr.2019.118564. Epub 2019 Oct 28. Biochim Biophys Acta Mol Cell Res. 2020. PMID: 31672612
-
[Epigenetic mechanisms and alcohol use disorders: a potential therapeutic target].Biol Aujourdhui. 2017;211(1):83-91. doi: 10.1051/jbio/2017014. Epub 2017 Jul 6. Biol Aujourdhui. 2017. PMID: 28682229 Review. French.
Cited by
-
Epigenetic Regulation of Multidrug Resistance Protein 1 and Breast Cancer Resistance Protein Transporters by Histone Deacetylase Inhibition.Drug Metab Dispos. 2020 Jun;48(6):459-480. doi: 10.1124/dmd.119.089953. Epub 2020 Mar 19. Drug Metab Dispos. 2020. PMID: 32193359 Free PMC article. Review.
-
Prenatal stress-induced disruptions in microbial and host tryptophan metabolism and transport.Behav Brain Res. 2021 Sep 24;414:113471. doi: 10.1016/j.bbr.2021.113471. Epub 2021 Jul 16. Behav Brain Res. 2021. PMID: 34280459 Free PMC article.
-
Histone Deacetylases (HDACs) and Atherosclerosis: A Mechanistic and Pharmacological Review.Front Cell Dev Biol. 2020 Nov 12;8:581015. doi: 10.3389/fcell.2020.581015. eCollection 2020. Front Cell Dev Biol. 2020. PMID: 33282862 Free PMC article. Review.
-
Epigenetic Effects Mediated by Antiepileptic Drugs and their Potential Application.Curr Neuropharmacol. 2020;18(2):153-166. doi: 10.2174/1570159X17666191010094849. Curr Neuropharmacol. 2020. PMID: 31660836 Free PMC article. Review.
-
Aryl hydrocarbon receptor: Its roles in physiology.Biochem Pharmacol. 2021 Mar;185:114428. doi: 10.1016/j.bcp.2021.114428. Epub 2021 Jan 28. Biochem Pharmacol. 2021. PMID: 33515530 Free PMC article. Review.
References
-
- Abbott NJ (2014) Anatomy and Physiology of the Blood-Brain Barriers In: Hammarlund-Udenaes M, de Lange E, Thorne RG (eds) Drug Delivery to the Brain: Physiogial Concepts, Methodologies and Approaches, vol 101 edn. Springer-Verlag; New York, pp xx, 731
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
