

Estimating the total genome length of a metagenomic sample using k-mers
- PMID: 30967110
- PMCID: PMC6456951
- DOI: 10.1186/s12864-019-5467-x
Estimating the total genome length of a metagenomic sample using k-mers
Abstract
Background: Metagenomic sequencing is a powerful technology for studying the mixture of microbes or the microbiomes on human and in the environment. One basic task of analyzing metagenomic data is to identify the component genomes in the community. This task is challenging due to the complexity of microbiome composition, limited availability of known reference genomes, and usually insufficient sequencing coverage.
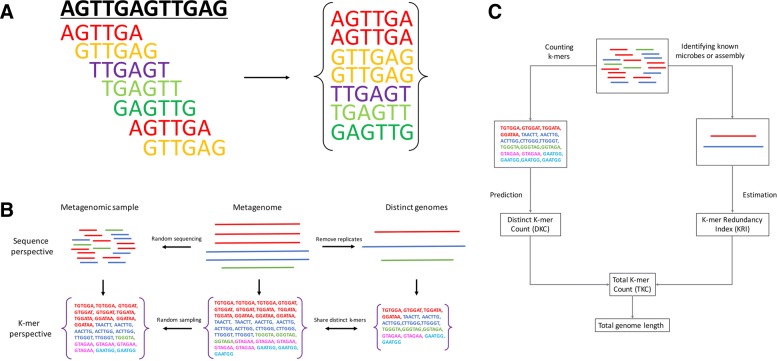
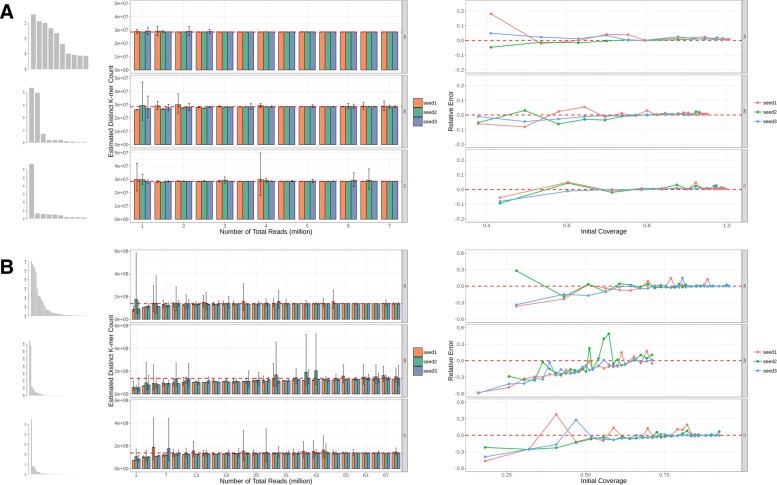
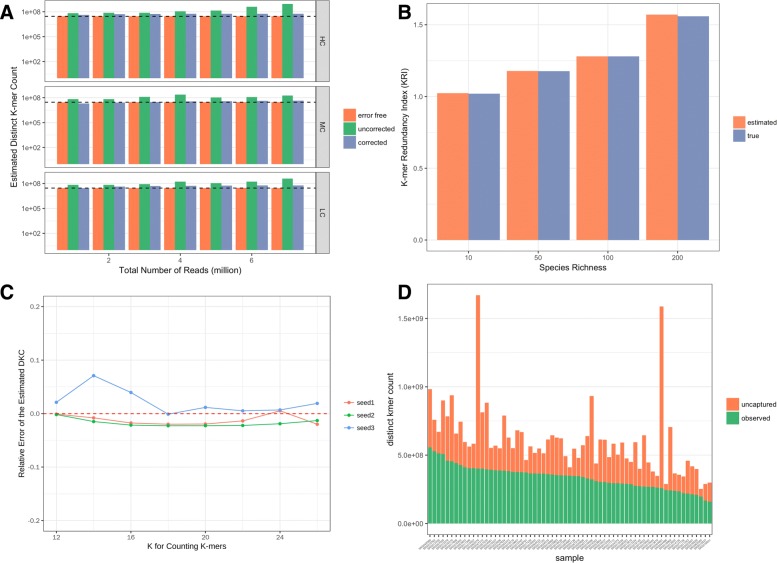
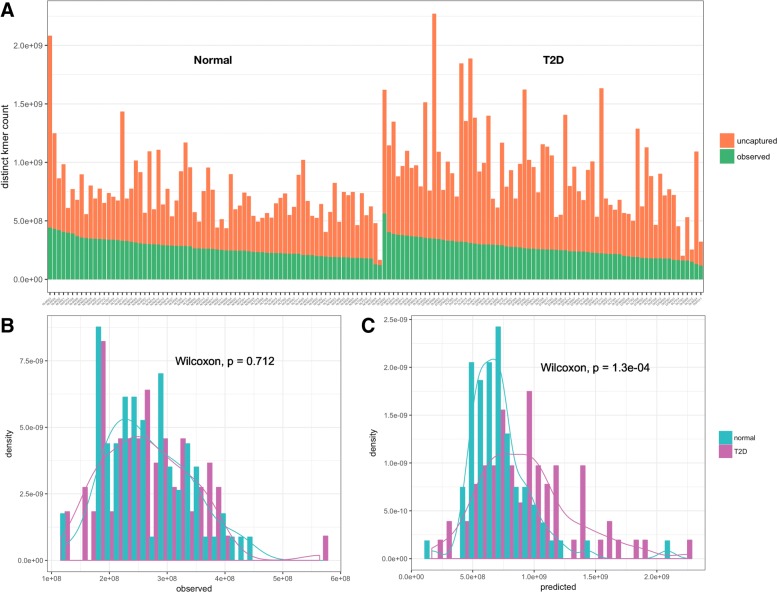
Results: As an initial step toward understanding the complete composition of a metagenomic sample, we studied the problem of estimating the total length of all distinct component genomes in a metagenomic sample. We showed that this problem can be solved by estimating the total number of distinct k-mers in all the metagenomic sequencing data. We proposed a method for this estimation based on the sequencing coverage distribution of observed k-mers, and introduced a k-mer redundancy index (KRI) to fill in the gap between the count of distinct k-mers and the total genome length. We showed the effectiveness of the proposed method on a set of carefully designed simulation data corresponding to multiple situations of true metagenomic data. Results on real data indicate that the uncaptured genomic information can vary dramatically across metagenomic samples, with the potential to mislead downstream analyses.
Conclusions: We proposed the question of how long the total genome length of all different species in a microbial community is and introduced a method to answer it.
Keywords: Distinct k-mers; Genome length; Metagenomics; Sequencing coverage.
Conflict of interest statement
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Figures
Similar articles
-
Assessment of k-mer spectrum applicability for metagenomic dissimilarity analysis.BMC Bioinformatics. 2016 Jan 16;17:38. doi: 10.1186/s12859-015-0875-7. BMC Bioinformatics. 2016. PMID: 26774270 Free PMC article.
-
Exploiting topic modeling to boost metagenomic reads binning.BMC Bioinformatics. 2015;16 Suppl 5(Suppl 5):S2. doi: 10.1186/1471-2105-16-S5-S2. Epub 2015 Mar 18. BMC Bioinformatics. 2015. PMID: 25859745 Free PMC article.
-
MetaCon: unsupervised clustering of metagenomic contigs with probabilistic k-mers statistics and coverage.BMC Bioinformatics. 2019 Nov 22;20(Suppl 9):367. doi: 10.1186/s12859-019-2904-4. BMC Bioinformatics. 2019. PMID: 31757198 Free PMC article.
-
Recovering metagenome-assembled genomes from shotgun metagenomic sequencing data: Methods, applications, challenges, and opportunities.Microbiol Res. 2022 Jul;260:127023. doi: 10.1016/j.micres.2022.127023. Epub 2022 Apr 8. Microbiol Res. 2022. PMID: 35430490 Review.
-
Metagenome analysis using the Kraken software suite.Nat Protoc. 2022 Dec;17(12):2815-2839. doi: 10.1038/s41596-022-00738-y. Epub 2022 Sep 28. Nat Protoc. 2022. PMID: 36171387 Free PMC article. Review.
Cited by
-
Enhancing Clinical Utility: Utilization of International Standards and Guidelines for Metagenomic Sequencing in Infectious Disease Diagnosis.Int J Mol Sci. 2024 Mar 15;25(6):3333. doi: 10.3390/ijms25063333. Int J Mol Sci. 2024. PMID: 38542307 Free PMC article. Review.
References
-
- Cui H, Li Y, Zhang X. An overview of major metagenomic studies on human microbiomes in health and disease. Quant Biol. 2016;4(3):192–206. doi: 10.1007/s40484-016-0078-x. - DOI
-
- Zhang X, Liu S, Cui H, Chen T. Reading the underlying information from massive metagenomic sequencing data. Proc IEEE. 2017;105(3):459–73.
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous
