

Global identification of functional microRNA-mRNA interactions in Drosophila
- PMID: 30967537
- PMCID: PMC6456604
- DOI: 10.1038/s41467-019-09586-z
Global identification of functional microRNA-mRNA interactions in Drosophila
Abstract
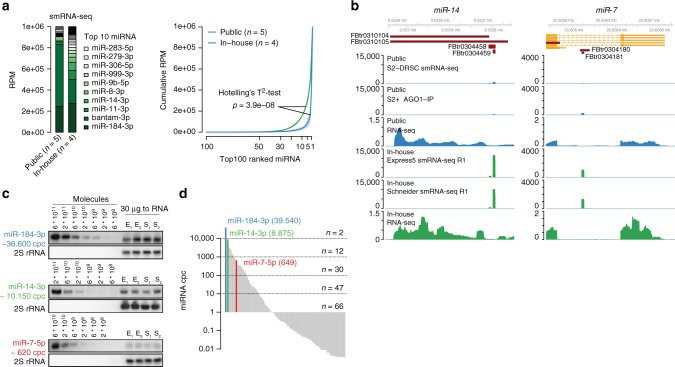
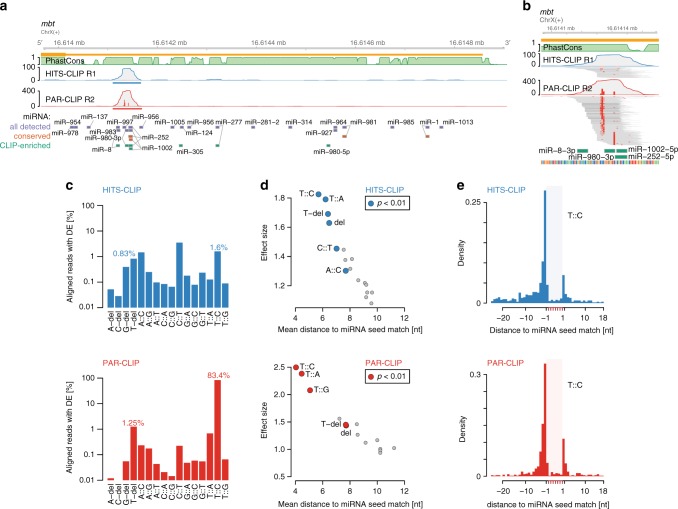
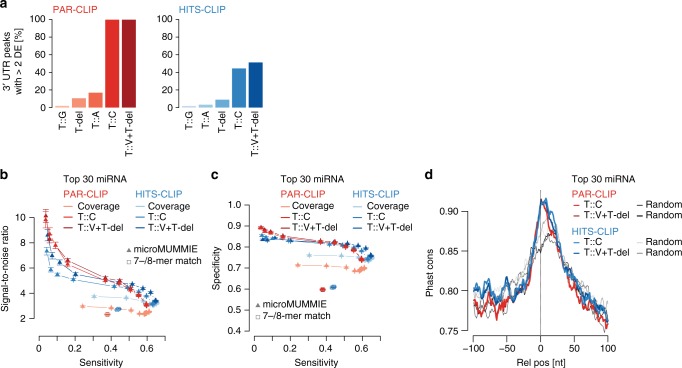
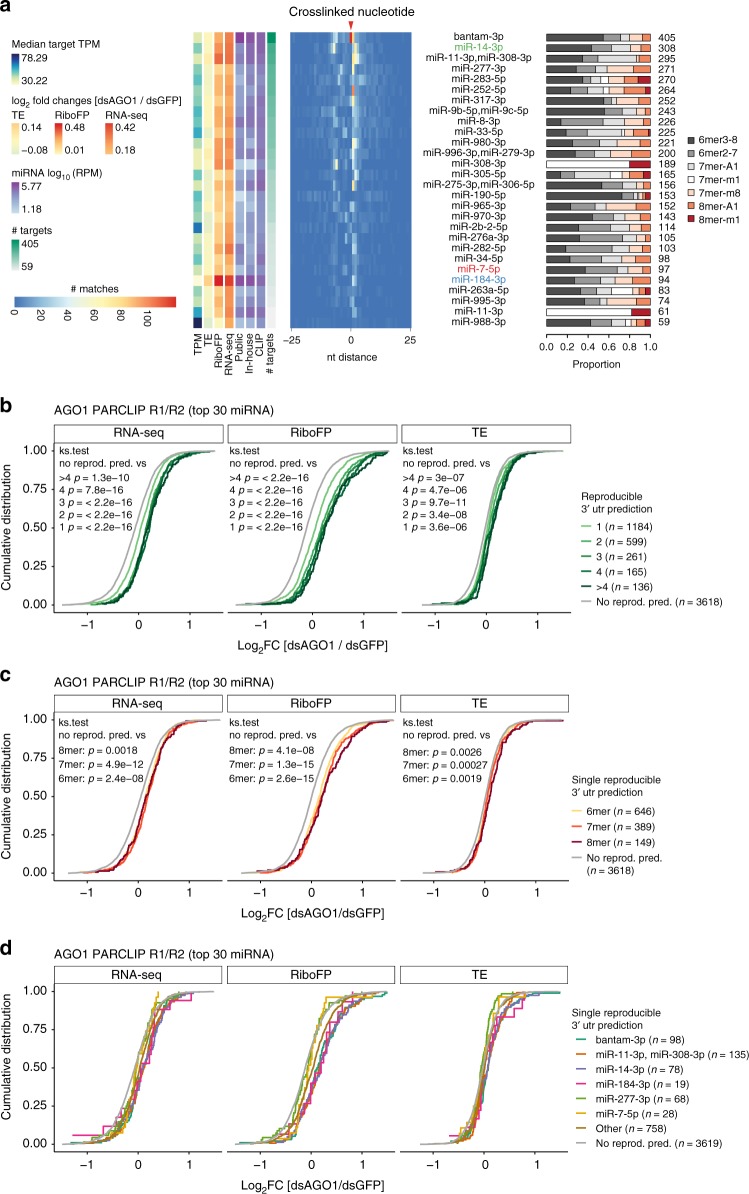
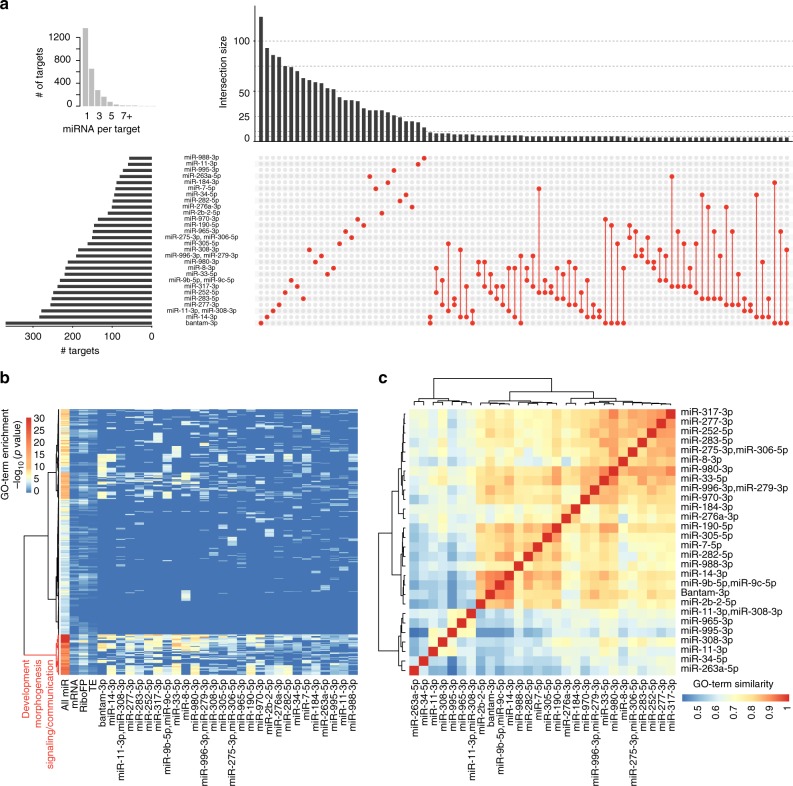
MicroRNAs (miRNAs) are key mediators of post-transcriptional gene expression silencing. So far, no comprehensive experimental annotation of functional miRNA target sites exists in Drosophila. Here, we generated a transcriptome-wide in vivo map of miRNA-mRNA interactions in Drosophila melanogaster, making use of single nucleotide resolution in Argonaute1 (AGO1) crosslinking and immunoprecipitation (CLIP) data. Absolute quantification of cellular miRNA levels presents the miRNA pool in Drosophila cell lines to be more diverse than previously reported. Benchmarking two CLIP approaches, we identify a similar predictive potential to unambiguously assign thousands of miRNA-mRNA pairs from AGO1 interaction data at unprecedented depth, achieving higher signal-to-noise ratios than with computational methods alone. Quantitative RNA-seq and sub-codon resolution ribosomal footprinting data upon AGO1 depletion enabled the determination of miRNA-mediated effects on target expression and translation. We thus provide the first comprehensive resource of miRNA target sites and their quantitative functional impact in Drosophila.
Conflict of interest statement
The authors declare no competing interests.
Figures
Similar articles
-
SmD1 Modulates the miRNA Pathway Independently of Its Pre-mRNA Splicing Function.PLoS Genet. 2015 Aug 26;11(8):e1005475. doi: 10.1371/journal.pgen.1005475. eCollection 2015 Aug. PLoS Genet. 2015. PMID: 26308709 Free PMC article.
-
Characterization of the miRNA-RISC loading complex and miRNA-RISC formed in the Drosophila miRNA pathway.RNA. 2009 Jul;15(7):1282-91. doi: 10.1261/rna.1541209. Epub 2009 May 18. RNA. 2009. PMID: 19451544 Free PMC article.
-
The 3'-to-5' exoribonuclease Nibbler shapes the 3' ends of microRNAs bound to Drosophila Argonaute1.Curr Biol. 2011 Nov 22;21(22):1878-87. doi: 10.1016/j.cub.2011.09.034. Epub 2011 Nov 3. Curr Biol. 2011. PMID: 22055293 Free PMC article.
-
MicroRNAs silence gene expression by repressing protein expression and/or by promoting mRNA decay.Cold Spring Harb Symp Quant Biol. 2006;71:523-30. doi: 10.1101/sqb.2006.71.013. Cold Spring Harb Symp Quant Biol. 2006. PMID: 17381335 Review.
-
Identification and consequences of miRNA-target interactions--beyond repression of gene expression.Nat Rev Genet. 2014 Sep;15(9):599-612. doi: 10.1038/nrg3765. Epub 2014 Jul 15. Nat Rev Genet. 2014. PMID: 25022902 Review.
Cited by
-
Aedes aegypti miRNA-33 modulates permethrin induced toxicity by regulating VGSC transcripts.Sci Rep. 2021 Mar 31;11(1):7301. doi: 10.1038/s41598-021-86665-6. Sci Rep. 2021. PMID: 33790374 Free PMC article.
-
Drosophila miR-263b-5p controls wing developmental growth by targeting Akt.Anim Cells Syst (Seoul). 2024 Dec 24;29(1):35-45. doi: 10.1080/19768354.2024.2444366. eCollection 2025. Anim Cells Syst (Seoul). 2024. PMID: 39777023 Free PMC article.
-
Exosomes as mediators of intercellular crosstalk in metabolism.Cell Metab. 2021 Sep 7;33(9):1744-1762. doi: 10.1016/j.cmet.2021.08.006. Cell Metab. 2021. PMID: 34496230 Free PMC article. Review.
-
The Biological Roles of microRNAs in Drosophila Development.Insects. 2024 Jun 30;15(7):491. doi: 10.3390/insects15070491. Insects. 2024. PMID: 39057224 Free PMC article. Review.
-
Noncoding RNA Regulation of Hormonal and Metabolic Systems in the Fruit Fly Drosophila.Metabolites. 2023 Jan 19;13(2):152. doi: 10.3390/metabo13020152. Metabolites. 2023. PMID: 36837772 Free PMC article. Review.
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
