

SETD2 mutations confer chemoresistance in acute myeloid leukemia partly through altered cell cycle checkpoints
- PMID: 30967619
- PMCID: PMC6785365
- DOI: 10.1038/s41375-019-0456-2
SETD2 mutations confer chemoresistance in acute myeloid leukemia partly through altered cell cycle checkpoints
Abstract
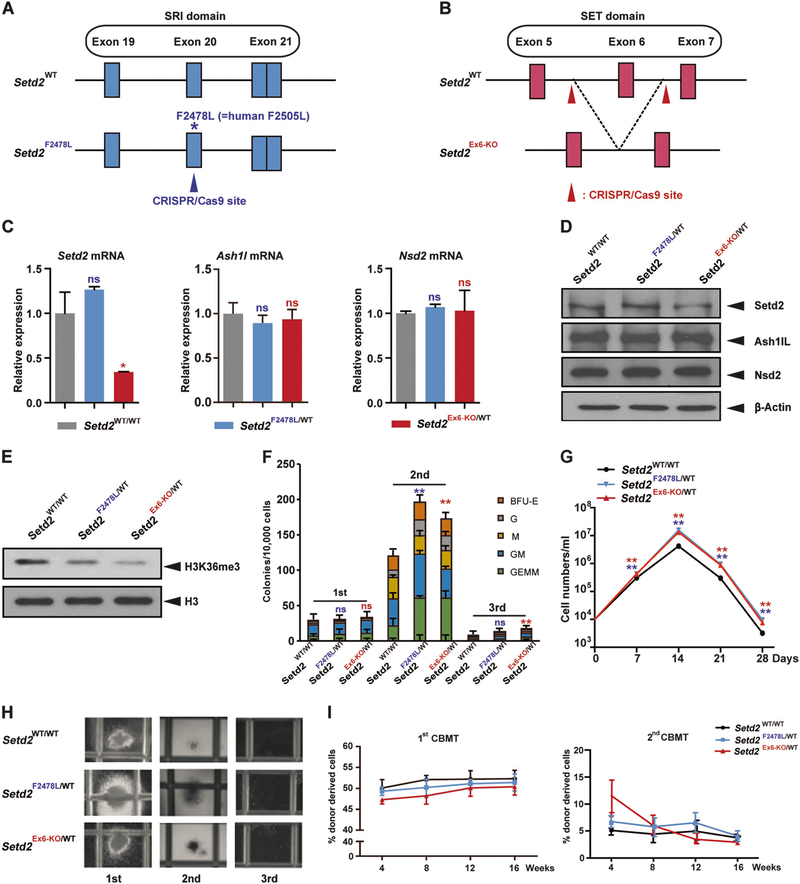
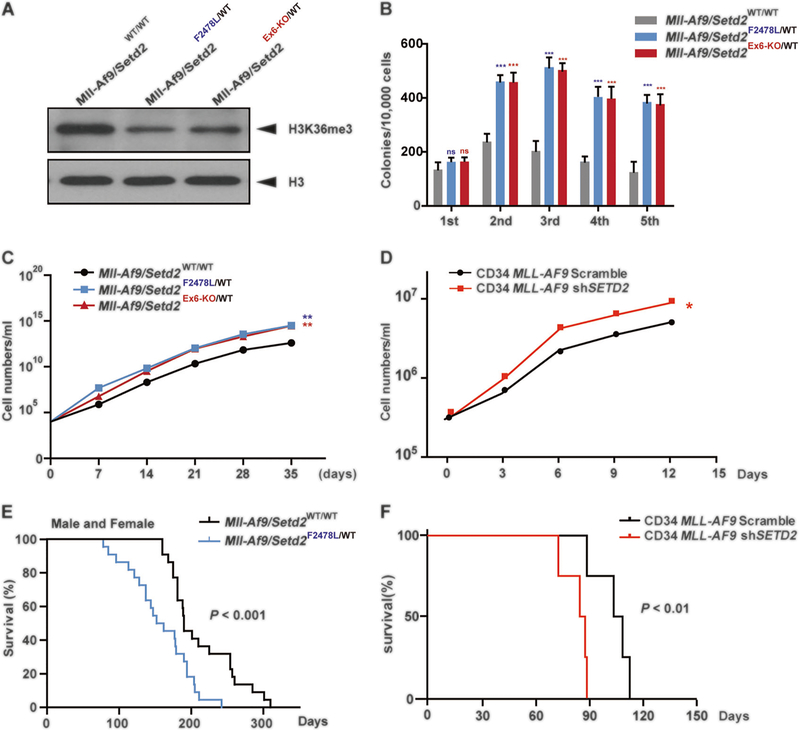
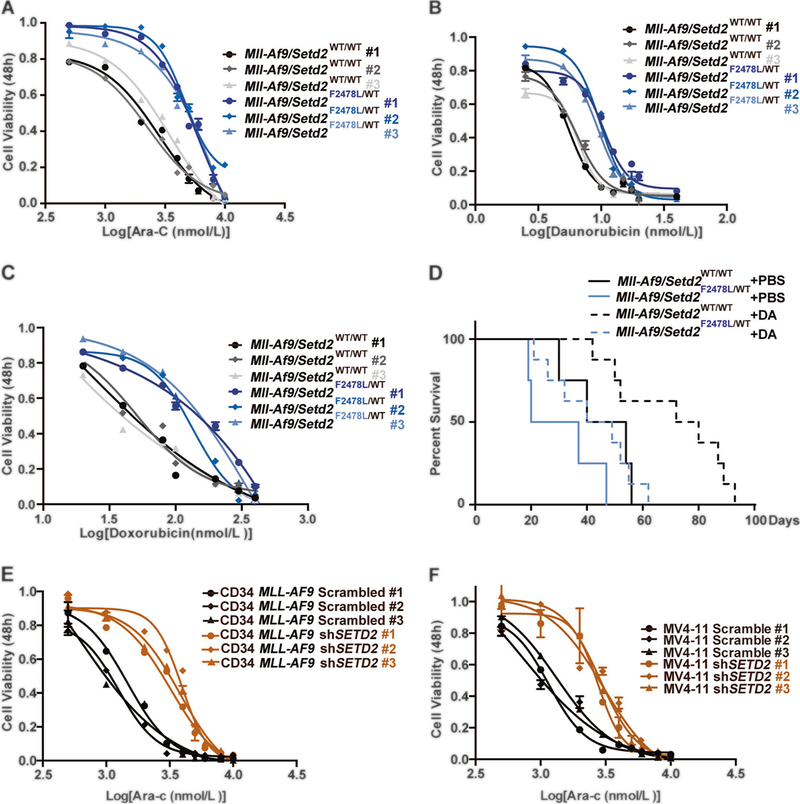
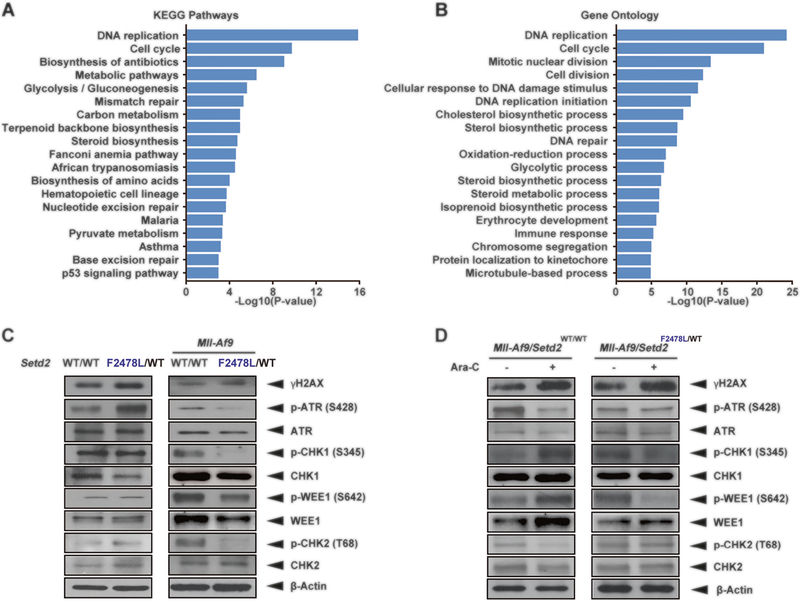
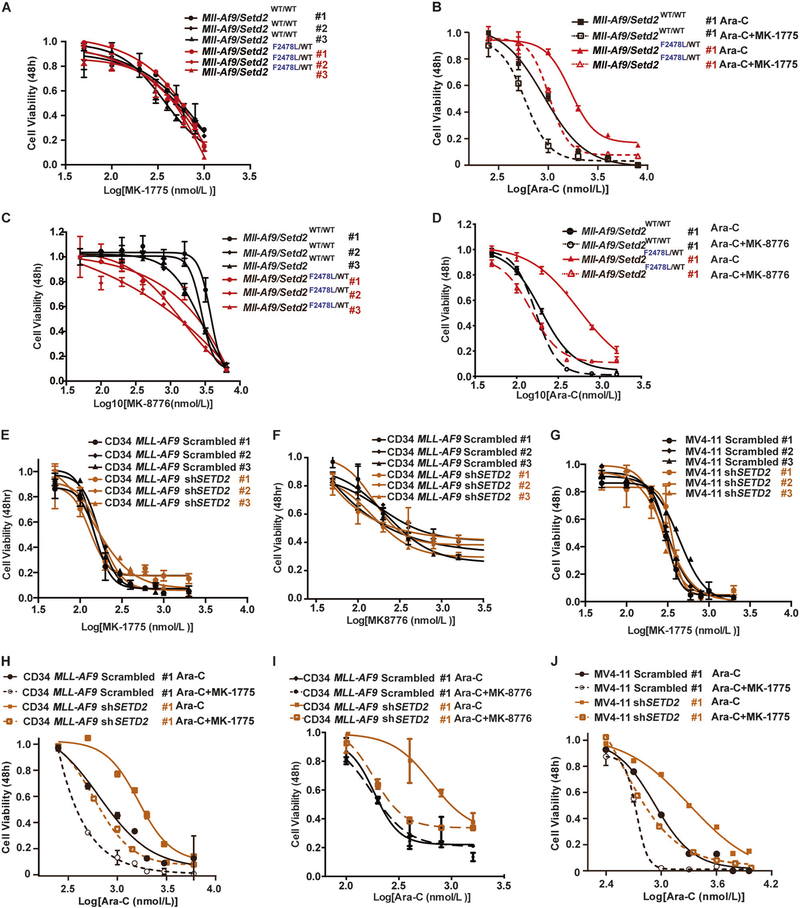
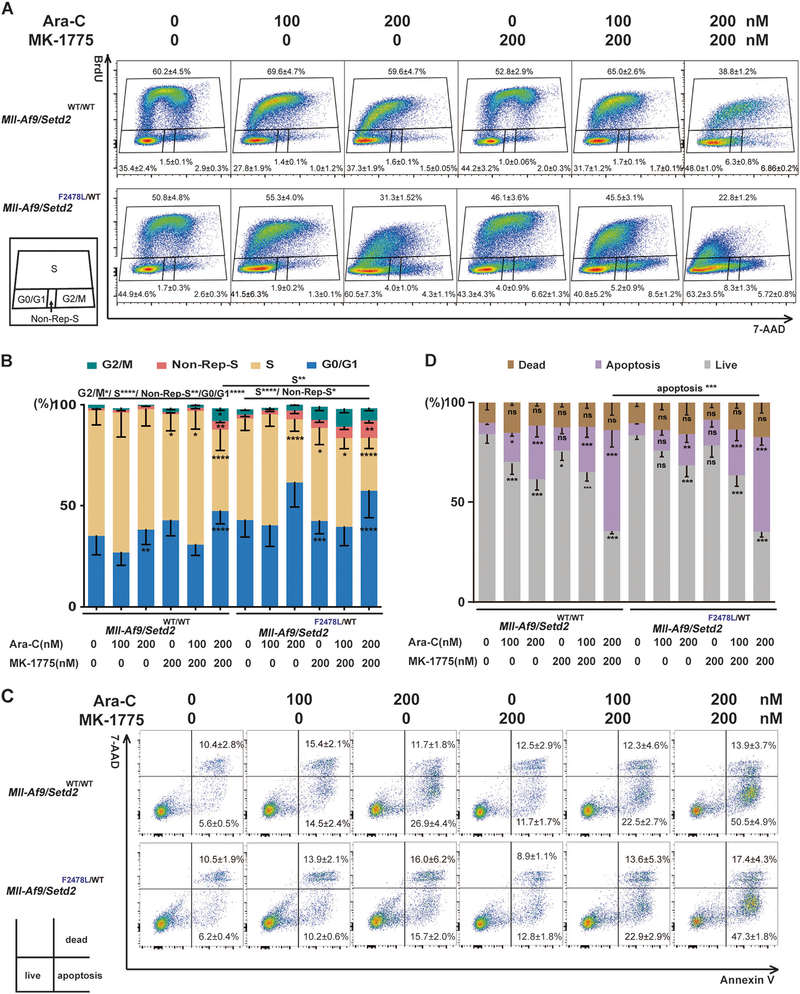
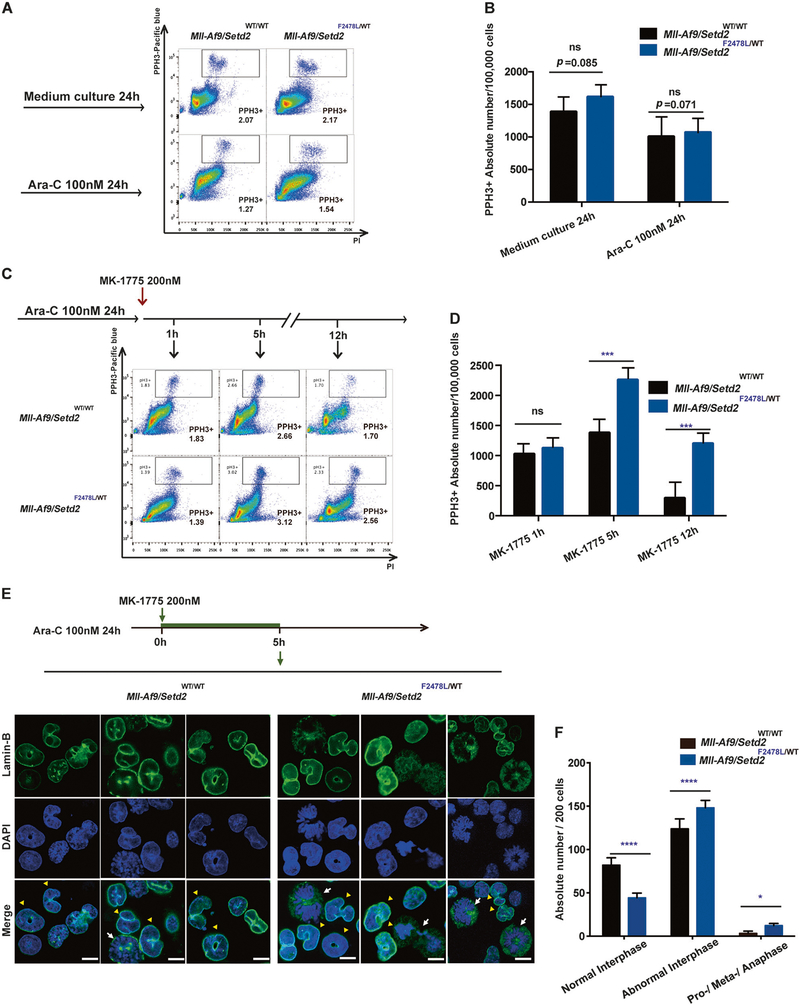
SETD2, an epigenetic tumor suppressor, is frequently mutated in MLL-rearranged (MLLr) leukemia and relapsed acute leukemia (AL). To clarify the impact of SETD2 mutations on chemotherapy sensitivity in MLLr leukemia, two loss-of-function (LOF) Setd2-mutant alleles (Setd2F2478L/WT or Setd2Ex6-KO/WT) were generated and introduced, respectively, to the Mll-Af9 knock-in leukemia mouse model. Both alleles cooperated with Mll-Af9 to accelerate leukemia development that resulted in resistance to standard Cytarabine-based chemotherapy. Mechanistically, Setd2-mutant leukemic cells showed downregulated signaling related to cell cycle progression, S, and G2/M checkpoint regulation. Thus, after Cytarabine treatment, Setd2-mutant leukemic cells exit from the S phase and progress to the G2/M phase. Importantly, S and G2/M cell cycle checkpoint inhibition could resensitize the Mll-Af9/Setd2 double-mutant cells to standard chemotherapy by causing DNA replication collapse, mitotic catastrophe, and increased cell death. These findings demonstrate that LOF SETD2 mutations confer chemoresistance on AL to DNA-damaging treatment by S and G2/M checkpoint defects. The combination of S and G2/M checkpoint inhibition with chemotherapy can be explored as a promising therapeutic strategy by exploiting their unique vulnerability and resensitizing chemoresistant AL with SETD2 or SETD2-like epigenetic mutations.
Conflict of interest statement
Figures
References
-
- Estey E, Dohner H. Acute myeloid leukaemia. Lancet. 2006;368:1894–907. - PubMed
-
- Short NJ, Ravandi F. Acute myeloid leukemia: past, present, and prospects for the future. Clin Lymphoma Myeloma Leuk. 2016;16 (Suppl):S25–9. - PubMed
-
- Nagler E, Xavier MF, Frey N. Updates in immunotherapy for acute myeloid leukemia. Transl Cancer Res. 2017;6:86–92.
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Research Materials
