

Glutaminase Deficiency Caused by Short Tandem Repeat Expansion in GLS
- PMID: 30970188
- PMCID: PMC8819703
- DOI: 10.1056/NEJMoa1806627
Glutaminase Deficiency Caused by Short Tandem Repeat Expansion in GLS
Abstract
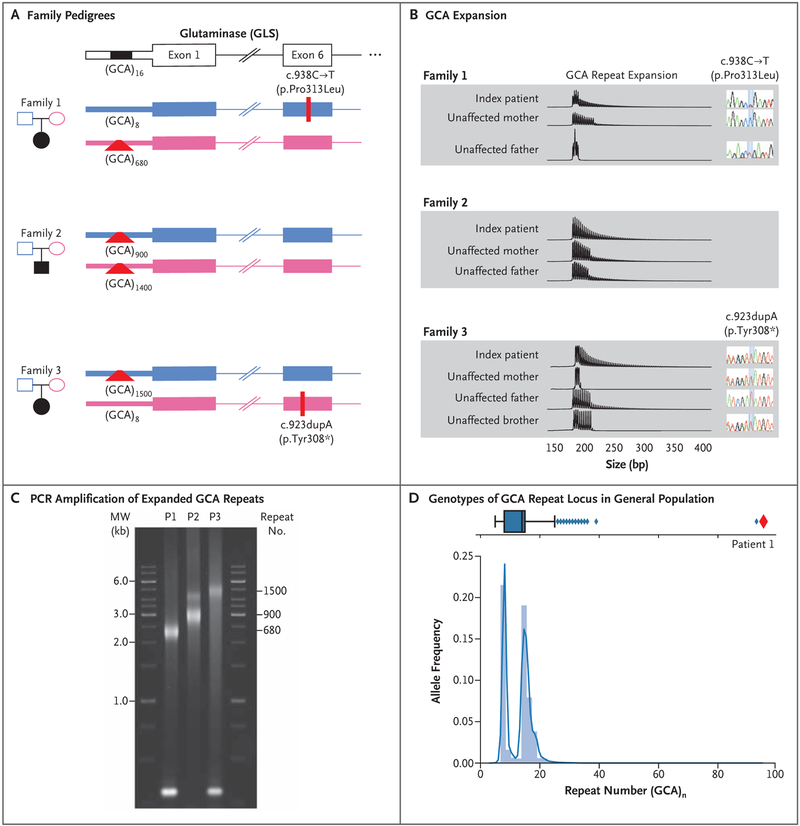
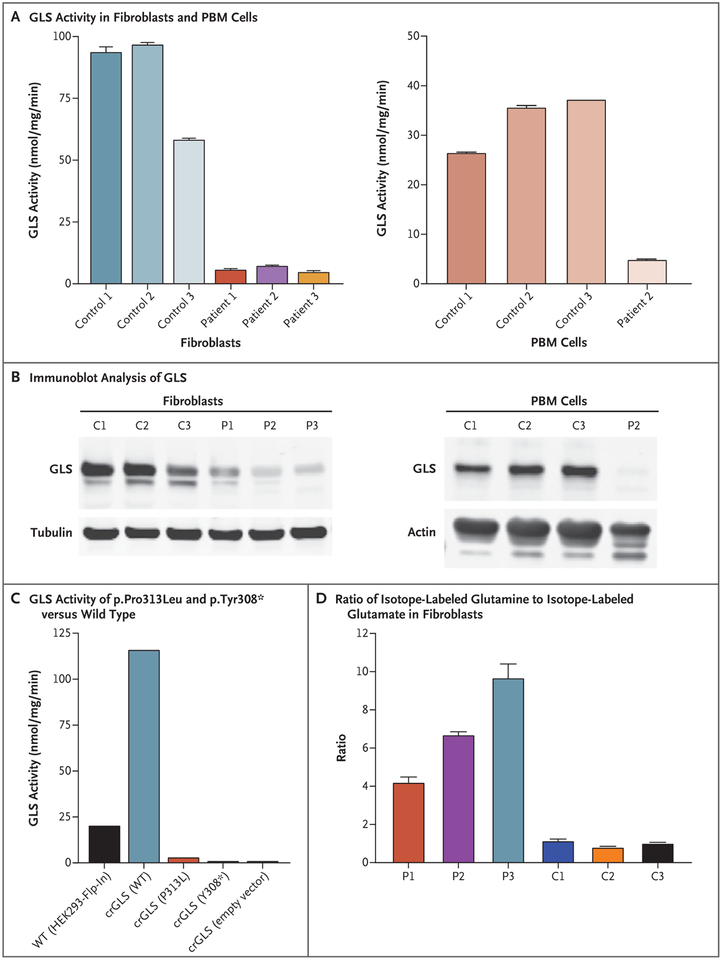
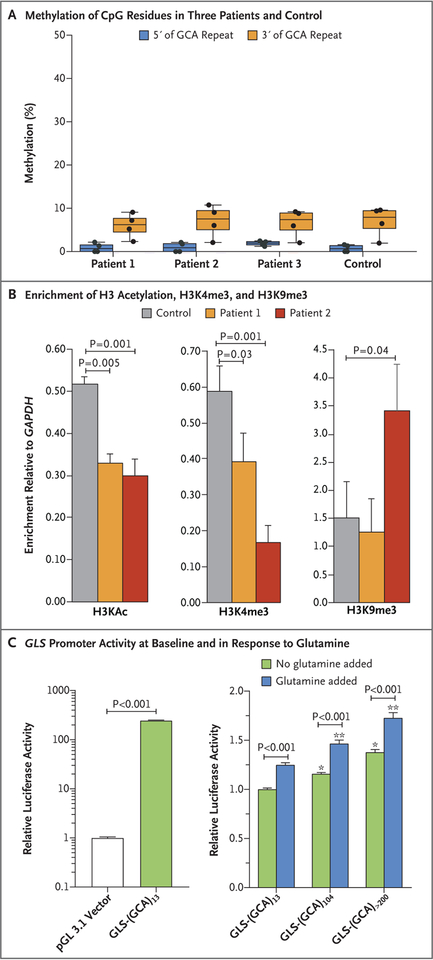
We report an inborn error of metabolism caused by an expansion of a GCA-repeat tract in the 5' untranslated region of the gene encoding glutaminase (GLS) that was identified through detailed clinical and biochemical phenotyping, combined with whole-genome sequencing. The expansion was observed in three unrelated patients who presented with an early-onset delay in overall development, progressive ataxia, and elevated levels of glutamine. In addition to ataxia, one patient also showed cerebellar atrophy. The expansion was associated with a relative deficiency of GLS messenger RNA transcribed from the expanded allele, which probably resulted from repeat-mediated chromatin changes upstream of the GLS repeat. Our discovery underscores the importance of careful examination of regions of the genome that are typically excluded from or poorly captured by exome sequencing.
Copyright © 2019 Massachusetts Medical Society.
Figures
Comment in
-
Glutaminase Deficiency Caused by Short Tandem Repeat Expansion in GLS.N Engl J Med. 2019 Sep 19;381(12):1185. doi: 10.1056/NEJMc1907427. N Engl J Med. 2019. PMID: 31532978 No abstract available.
References
-
- Wright CF, FitzPatrick DR, Firth HV. Paediatric genomics: diagnosing rare disease in children. Nat Rev Genet 2018; 19: 253–68. - PubMed
-
- Gilissen C, Hehir-Kwa JY, Thung DT, et al. Genome sequencing identifies major causes of severe intellectual disability. Nature 2014; 511: 344–7. - PubMed
-
- Alfares A, Aloraini T, Subaie LA, et al. Whole-genome sequencing offers additional but limited clinical utility compared with reanalysis of whole-exome sequencing. Genet Med 2018; 20: 1328–33. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Miscellaneous