

Mechanisms of B-Cell Oncogenesis Induced by Epstein-Barr Virus
- PMID: 30971472
- PMCID: PMC6580952
- DOI: 10.1128/JVI.00238-19
Mechanisms of B-Cell Oncogenesis Induced by Epstein-Barr Virus
Abstract
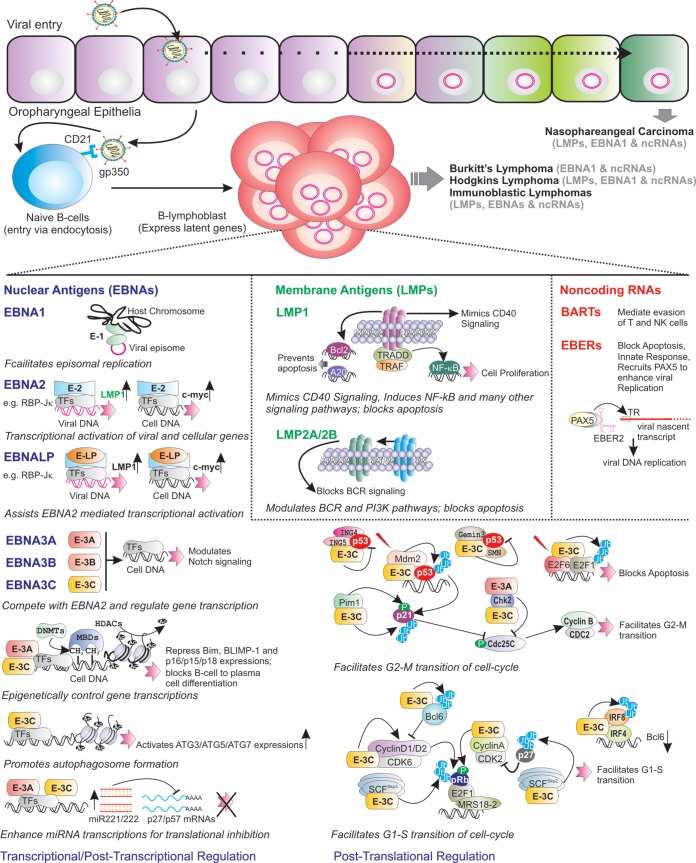
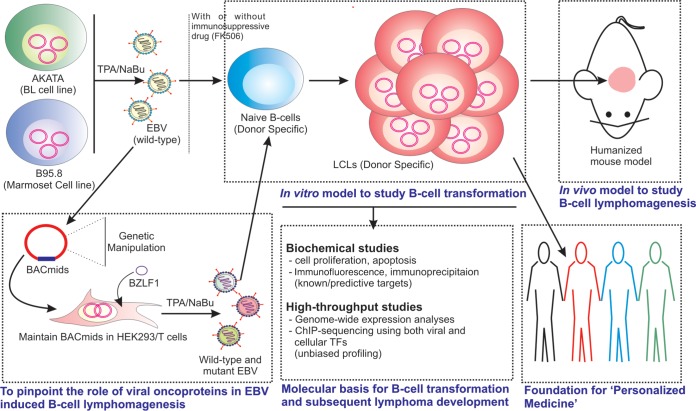
Epstein-Barr virus (EBV) is a ubiquitous gammaherpesvirus which asymptomatically infects the majority of the world population. Under immunocompromised conditions, EBV can trigger human cancers of epithelial and lymphoid origin. The oncogenic potential of EBV is demonstrated by in vitro infection and transformation of quiescent B cells into lymphoblastoid cell lines (LCLs). These cell lines, along with primary infection using genetically engineered viral particles coupled with recent technological advancements, have elucidated the underlying mechanisms of EBV-induced B-cell lymphomagenesis.
Keywords: B-cell lymphomas; Epstein-Barr virus; lymphoblastoid cell lines; tumor virology.
Copyright © 2019 Saha and Robertson.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
