

Physicochemical Properties, Biotransformation, and Transport Pathways of Established and Newly Approved Medications: A Systematic Review of the Top 200 Most Prescribed Drugs vs. the FDA-Approved Drugs Between 2005 and 2016
- PMID: 30972694
- PMCID: PMC6773482
- DOI: 10.1007/s40262-019-00750-8
Physicochemical Properties, Biotransformation, and Transport Pathways of Established and Newly Approved Medications: A Systematic Review of the Top 200 Most Prescribed Drugs vs. the FDA-Approved Drugs Between 2005 and 2016
Abstract
Background: Enzyme-mediated biotransformation of pharmacological agents is a crucial step in xenobiotic detoxification and drug disposition. Herein, we investigated the metabolism and physicochemical properties of the top 200 most prescribed drugs (established) as well as drugs approved by the US Food and Drug Administration (FDA) between 2005 and 2016 (newly approved).
Objective: Our objective was to capture the changing trends in the routes of administration, physicochemical properties, and prodrug medications, as well as the contributions of drug-metabolizing enzymes and transporters to drug clearance.
Methods: The University of Washington Drug Interaction Database (DIDB®) as well as other online resources (e.g., CenterWatch.com, Drugs.com, DrugBank.ca, and PubChem.ncbi.nlm.nih.gov) was used to collect and stratify the dataset required for exploring the above-mentioned trends.
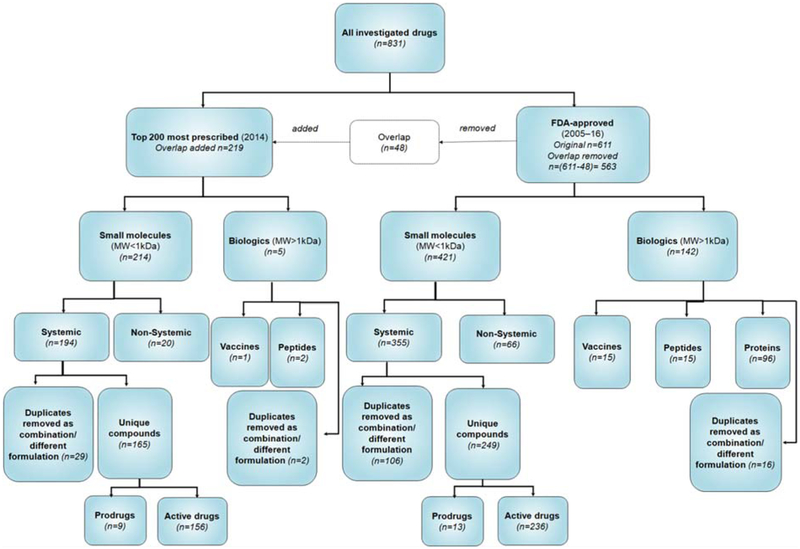
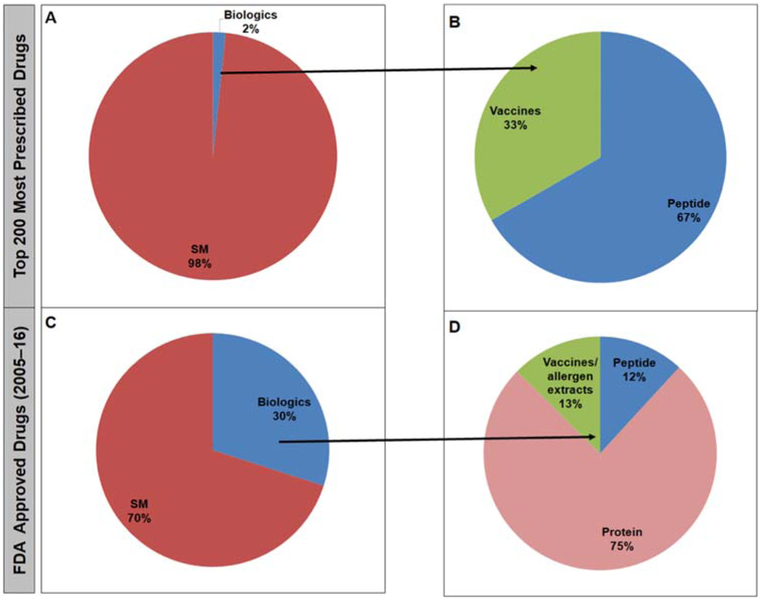
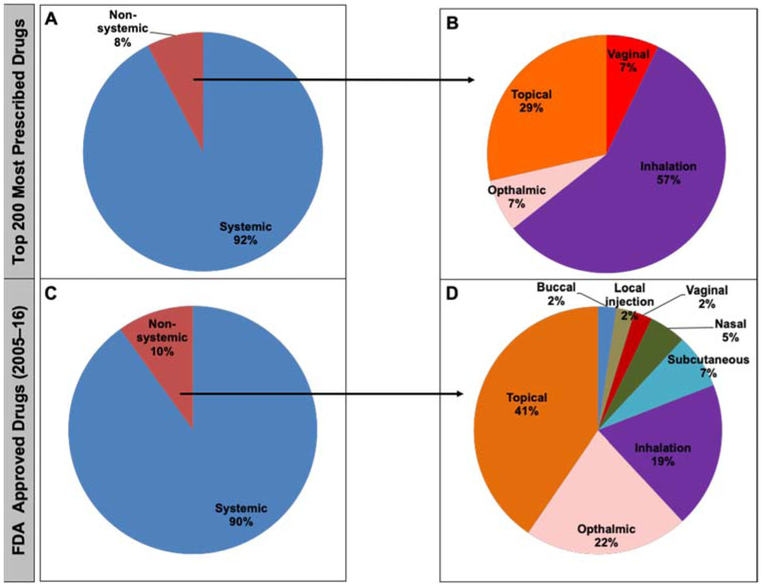
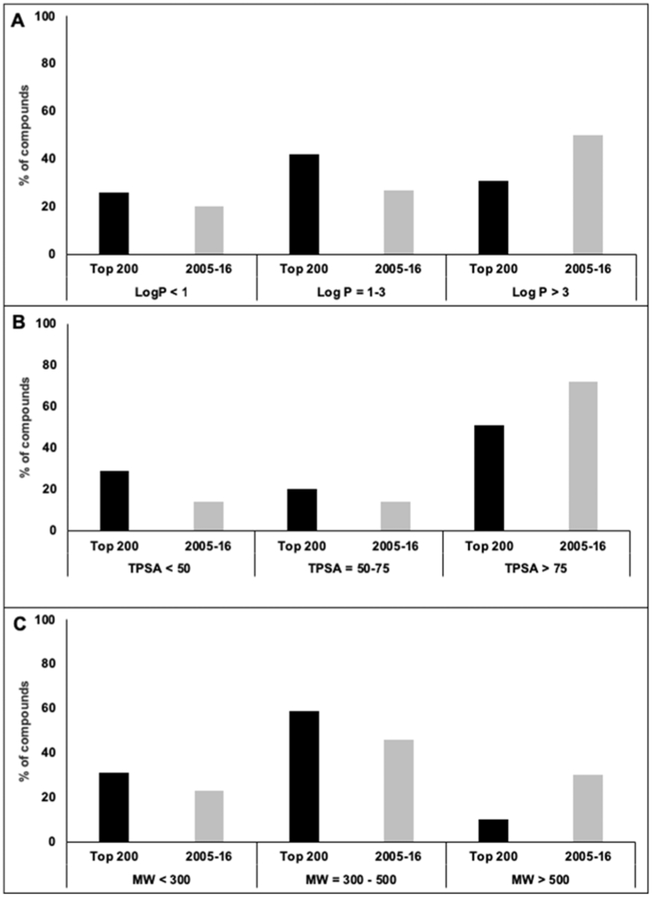
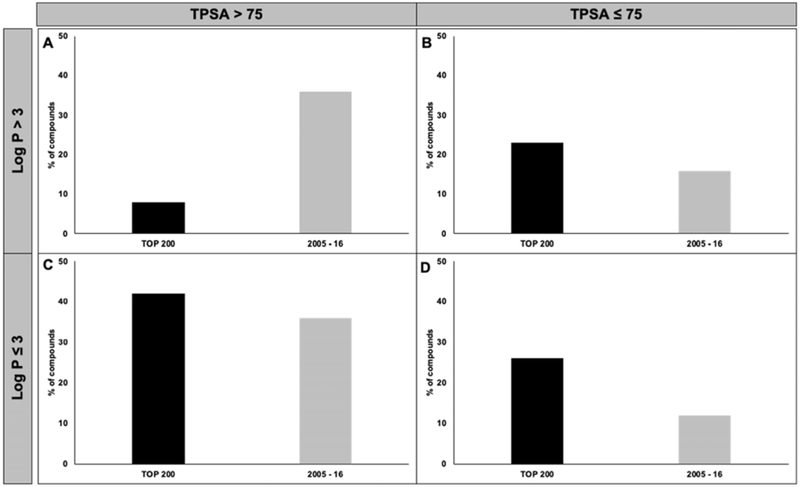
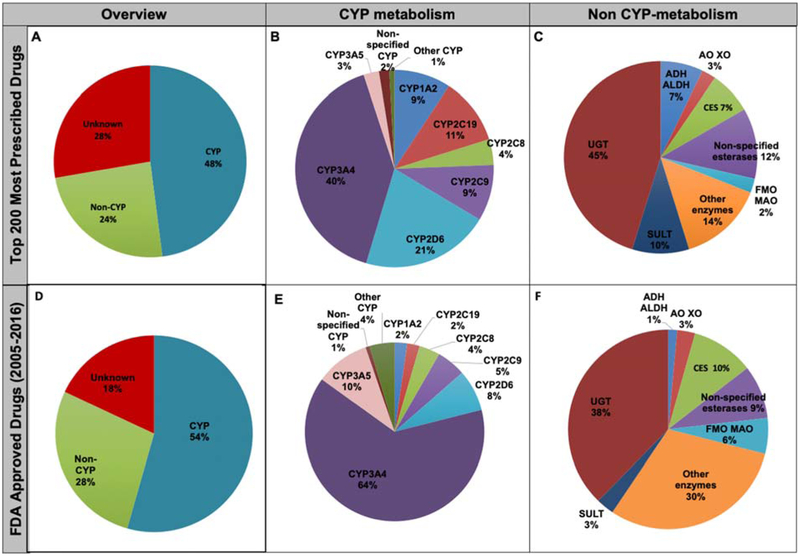
Results: Analyses revealed that ~ 90% of all drugs in the established and newly approved drug lists were administered systemically (oral or intravenous). Meanwhile, the portion of biologics (molecular weight > 1 kDa) was 15 times greater in the newly approved list than established drugs. Additionally, there was a 4.5-fold increase in the number of compounds with a high calculated partition coefficient (cLogP > 3) and a high total polar surface area (> 75 Å2) in the newly approved drug vs. the established category. Further, prodrugs in established or newly approved lists were found to be converted to active compounds via hydrolysis, demethylases, and kinases. The contribution of cytochrome P450 (CYP) 3A4, as the major biotransformation pathway, has increased from 40% in the established drug list to 64% in the newly approved drug list. Moreover, the role of CYP1A2, CYP2C19, and CYP2D6 were decreased as major metabolizing enzymes among the newly approved medications. Among non-CYP major metabolizers, the contribution of alcohol dehydrogenases/aldehyde dehydrogenases (ADH/ALDH) and sulfotransferases decreased in the newly approved drugs compared with the established list. Furthermore, the highest contribution among uptake and efflux transporters was found for Organic Anion Transporting Polypeptide 1B1 (OATP1B1) and P-glycoprotein (P-gp), respectively.
Conclusions: The higher portion of biologics in the newly approved drugs compared with the established list confirmed the growing demands for protein- and antibody-based therapies. Moreover, the larger number of hydrophilic drugs found in the newly approved list suggests that the probability of toxicity is likely to decrease. With regard to CYP-mediated major metabolism, CYP3A5 showed an increased involvement owing to the identification of unique probe substrates to differentiate CYP3As. Furthermore, the contribution of OATP1B1 and P-gp did not show a significant shift in the newly approved drugs as compared to the established list because of their broad substrate specificity.
Conflict of interest statement
Figures
References
-
- Oda S, Fukami T, Yokoi T, Nakajima M. A comprehensive review of UDP-glucuronosyltransferase and esterases for drug development. Drug Metab Pharmacokinet. 2015;30(1):30–51. - PubMed
-
- Iyanagi T Molecular mechanism of phase I and phase II drugmetabolizing enzymes: implications for detoxification. Int Rev Cytol. 2007;260:35–112. - PubMed
-
- Williams JA, Hyland R, Jones BC, Smith DA, Hurst S, Goosen TC, et al. Drug-drug interactions for UDP-glucuronosyltransferase substrates: a pharmacokinetic explanation for typically observed low exposure (AUCi/AUC) ratios. Drug Metab Dispos. 2004;32(11):1201–8. - PubMed
-
- Nelson DR, Zeldin DC, Hoffman SM, Maltais LJ, Wain HM, Nebert DW. Comparison of cytochrome P450 (CYP) genes from the mouse and human genomes, including nomenclature recommendations for genes, pseudogenes and alternative-splice variants. Pharmacogenetics. 2004;14(1):1–18. - PubMed
-
- Nebert DW, Russell DW. Clinical importance of the cytochromes P450. Lancet. 2002;360(9340):1155–62. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous
