

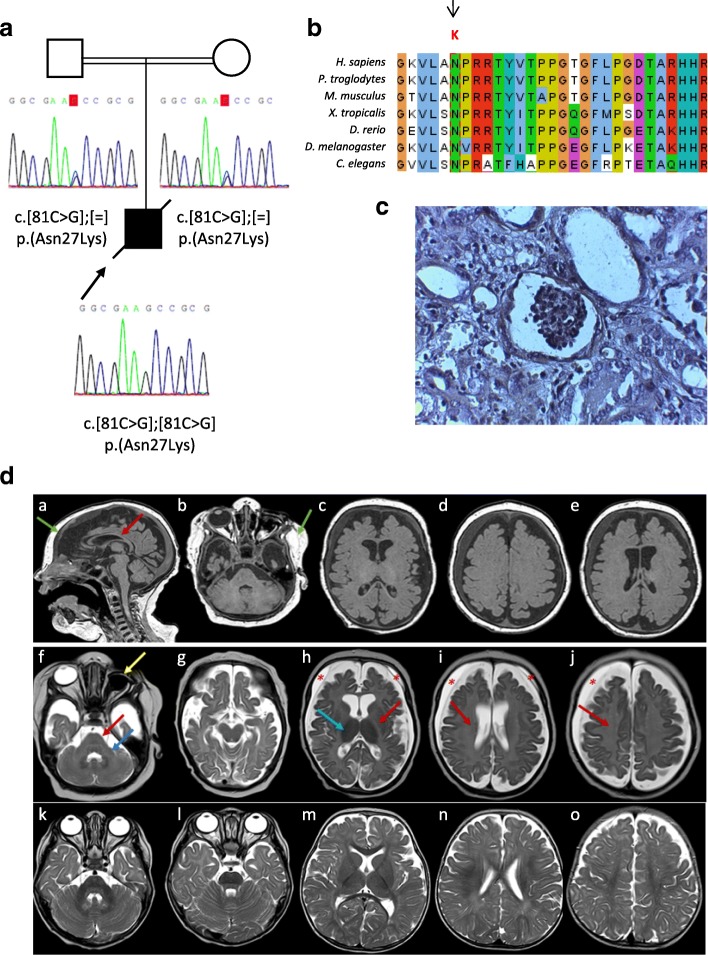
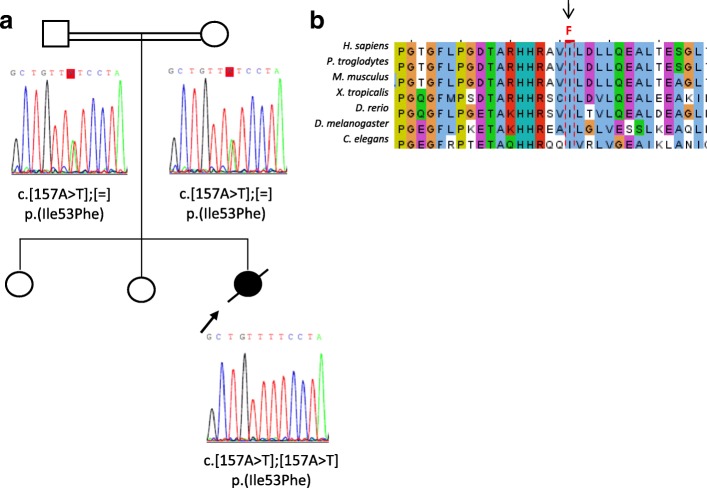
Novel homozygous OSGEP gene pathogenic variants in two unrelated patients with Galloway-Mowat syndrome: case report and review of the literature
- PMID: 30975089
- PMCID: PMC6458604
- DOI: 10.1186/s12882-019-1317-y
Novel homozygous OSGEP gene pathogenic variants in two unrelated patients with Galloway-Mowat syndrome: case report and review of the literature
Abstract
Background: Galloway-Mowat syndrome (GAMOS) is a rare autosomal recessive disorder characterized by early-onset nephrotic syndrome and microcephaly with brain anomalies. WDR73 pathogenic variants were described as the first genetic cause of GAMOS and, very recently, four novel causative genes, OSGEP, LAGE3, TP53RK, and TPRKB, have been identified.
Case presentation: We present the clinical and genetic characteristics of two unrelated infants with clinical suspicion of GAMOS who were born from consanguineous parents. Both patients showed a similar clinical presentation, with early-onset nephrotic syndrome, microcephaly, brain atrophy, developmental delay, axial hypotonia, and early fatality. We identified two novel likely disease-causing variants in the OSGEP gene. These two cases, in conjunction with the findings of a literature review, indicate that OSGEP pathogenic variants are associated with an earlier onset of nephrotic syndrome and shorter life expectancy than WDR73 pathogenic variants.
Conclusions: Our findings expand the spectrum of pathogenic variants in the OSGEP gene and, taken in conjunction with the results of the literature review, suggest that the OSGEP gene should be considered the main known monogenic cause of GAMOS. Early genetic diagnosis of GAMOS is of paramount importance for genetic counseling and family planning.
Keywords: Case report; Galloway-Mowat syndrome; Genetic testing; KEOPS complex; Nephrotic syndrome; OSGEP.
Conflict of interest statement
Ethics approval and consent to participate
This study was approved by Fundació Puigvert Institutional Review Board. The parents of the patients provided written informed consent to participate in this study.
Consent for publication
The parents of the patients provided written informed consent to publish this case report, including case description, medical data, and images, maintaining anonymity.
Competing interests
RT is the Editorial Board Member of BMC Nephrology. The other authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Figures
References
Publication types
MeSH terms
Substances
Supplementary concepts
LinkOut - more resources
Full Text Sources
Medical
