

Sensitive Quantitative Proteomics of Human Hematopoietic Stem and Progenitor Cells by Data-independent Acquisition Mass Spectrometry
- PMID: 30975897
- PMCID: PMC6601215
- DOI: 10.1074/mcp.TIR119.001431
Sensitive Quantitative Proteomics of Human Hematopoietic Stem and Progenitor Cells by Data-independent Acquisition Mass Spectrometry
Abstract
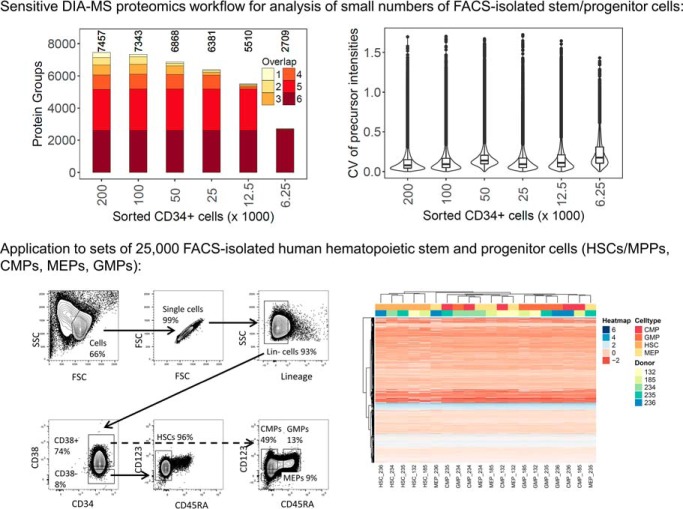
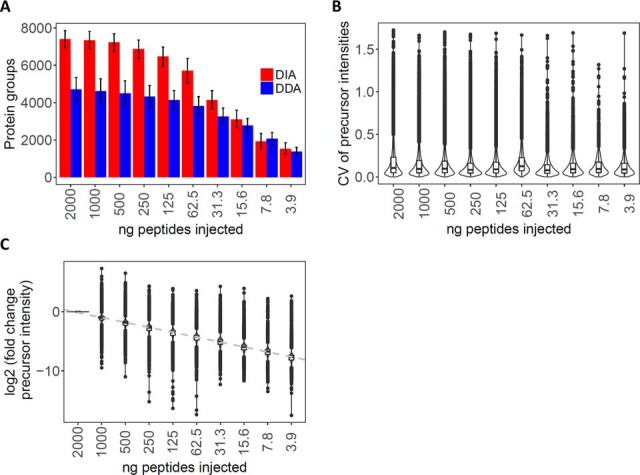
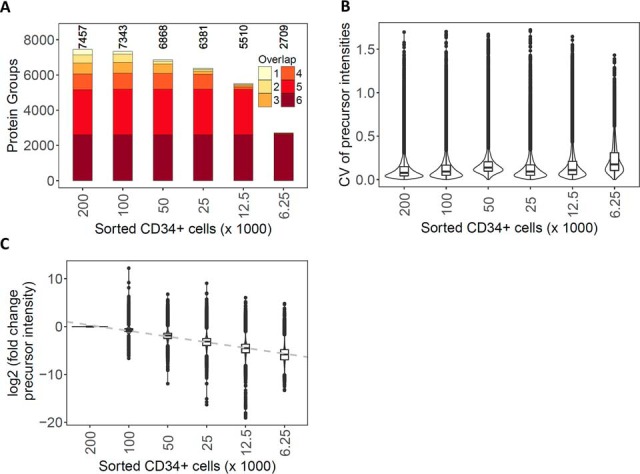
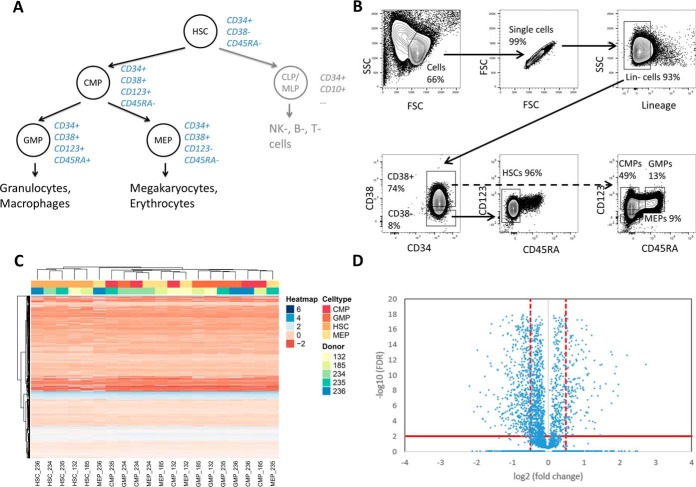
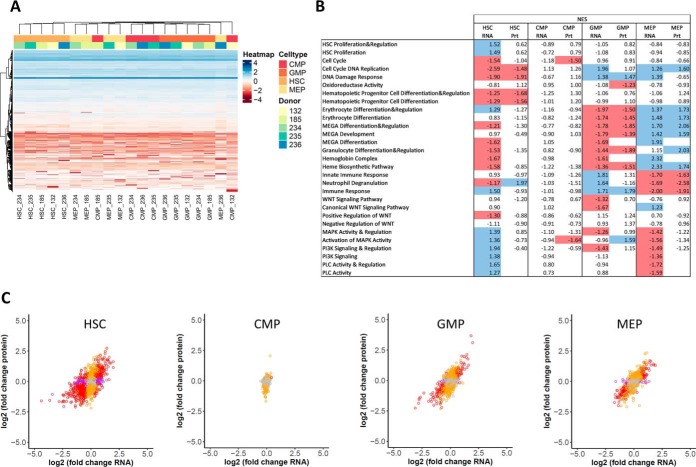
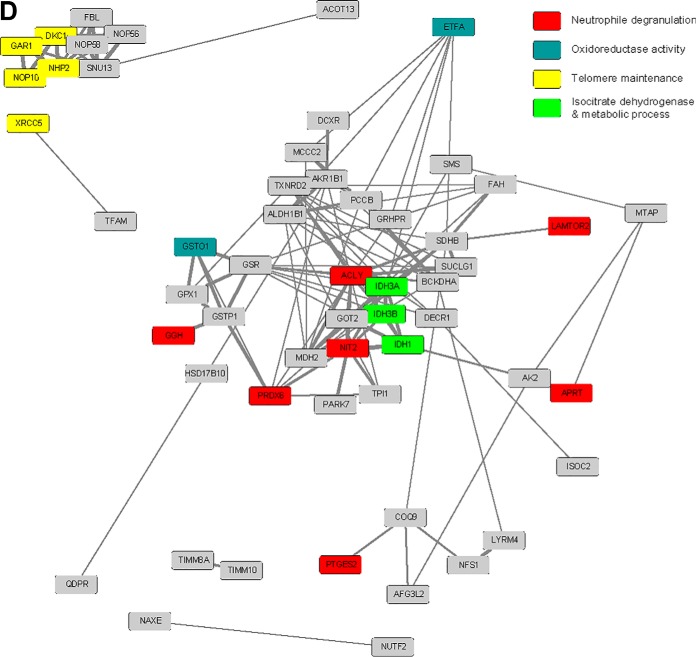
Physiological processes in multicellular organisms depend on the function and interactions of specialized cell types operating in context. Some of these cell types are rare and thus obtainable only in minute quantities. For example, tissue-specific stem and progenitor cells are numerically scarce, but functionally highly relevant, and fulfill critical roles in development, tissue maintenance, and disease. Whereas low numbers of cells are routinely analyzed by genomics and transcriptomics, corresponding proteomic analyses have so far not been possible due to methodological limitations. Here we describe a sensitive and robust quantitative technique based on data-independent acquisition mass spectrometry. We quantified the proteome of sets of 25,000 human hematopoietic stem/multipotent progenitor cells (HSC/MPP) and three committed progenitor cell subpopulations of the myeloid differentiation pathway (common myeloid progenitors, megakaryocyte-erythrocyte progenitors, and granulocyte-macrophage progenitors), isolated by fluorescence-activated cell sorting from five healthy donors. On average, 5,851 protein groups were identified per sample. A subset of 4,131 stringently filtered protein groups was quantitatively compared across the 20 samples, defining unique signatures for each subpopulation. A comparison of proteomic and transcriptomic profiles indicated HSC/MPP-specific divergent regulation of biochemical functions such as telomerase maintenance and quiescence-inducing enzymes, including isocitrate dehydrogenases. These are essential for maintaining stemness and were detected at proteome, but not transcriptome, level. The method is equally applicable to almost any rare cell type, including healthy and cancer stem cells or physiologically and pathologically infiltrating cell populations. It thus provides essential new information toward the detailed biochemical understanding of cell development and functionality in health and disease.
Keywords: Cell Sorting; Clinical Proteomics; Data-Independent Acquisition Mass Spectrometry; Differentiation*; Fluorescence-Activated Cell Sorting; Hematopoietic Stem and Progenitor Cells; Low Cell Number; Mass Spectrometry; Proteomics; Quantification.
© 2019 Amon et al.
Conflict of interest statement
R.A. holds shares of Biognosys AG, which operates in the field covered by the article. The remaining authors declare no competing financial interests
Figures
References
-
- Notta F., Zandi S., Takayama N., Dobson S., Gan O. I., Wilson G., Kaufmann K. B., McLeod J., Laurenti E., Dunant C. F., McPherson J. D., Stein L. D., Dror Y., and Dick J. E. (2016) Distinct routes of lineage development reshape the human blood hierarchy across ontogeny. Science 351, aab2116. - PMC - PubMed
-
- Shlush L. I., Zandi S., Mitchell A., Chen W. C., Brandwein J. M., Gupta V., Kennedy J. A., Schimmer A. D., Schuh A. C., Yee K. W., McLeod J. L., Doedens M., Medeiros J. J., Marke R., Kim H. J., Lee K., McPherson J. D., Hudson T. J., HALT Pan-Leukemia Gene Panel Consortium, Brown A. M., Yousif F., Trinh Q. M., Stein L. D., Minden M. D., Wang J. C., and Dick J. E. (2014) Identification of pre-leukaemic haematopoietic stem cells in acute leukaemia. Nature 506, 328–333 - PMC - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
