

Mechanisms of neurodegeneration in a preclinical autosomal dominant retinitis pigmentosa knock-in model with a RhoD190N mutation
- PMID: 30976840
- PMCID: PMC7144803
- DOI: 10.1007/s00018-019-03090-9
Mechanisms of neurodegeneration in a preclinical autosomal dominant retinitis pigmentosa knock-in model with a RhoD190N mutation
Abstract
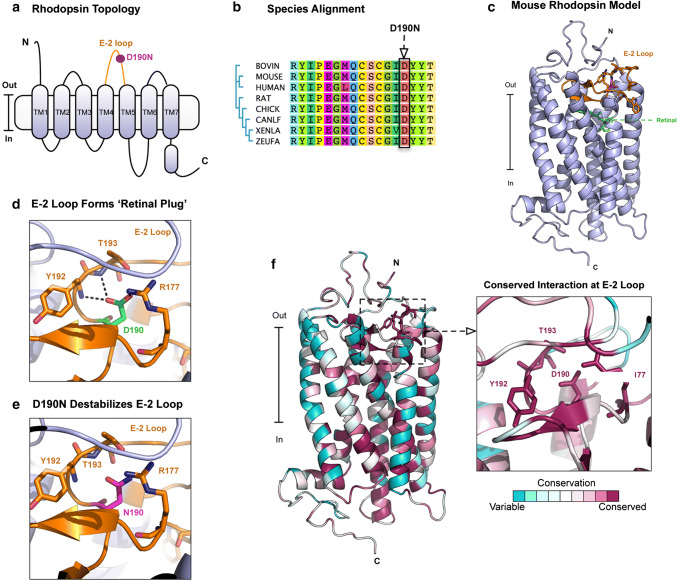
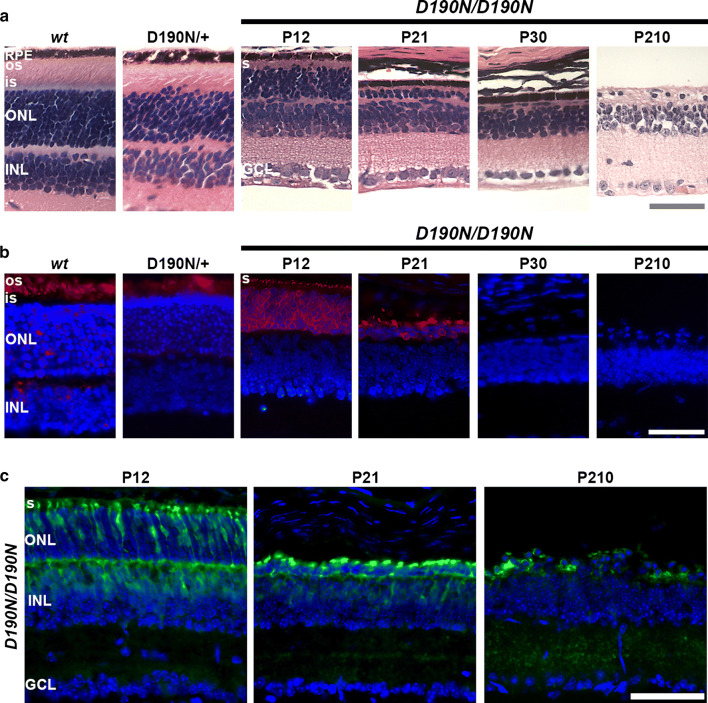
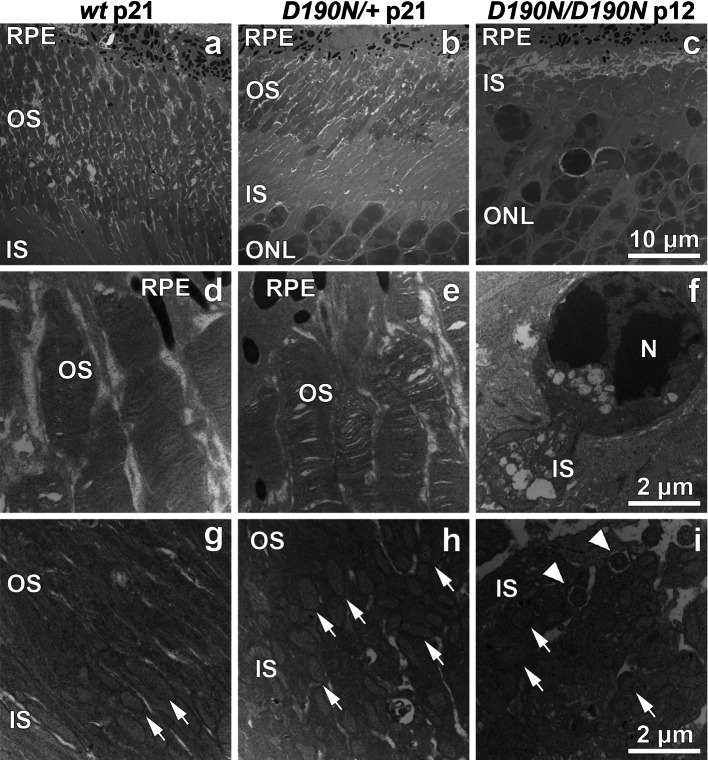
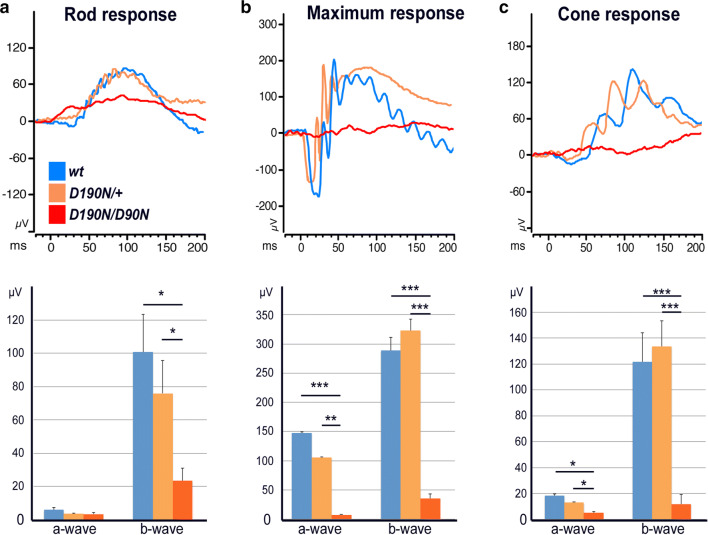
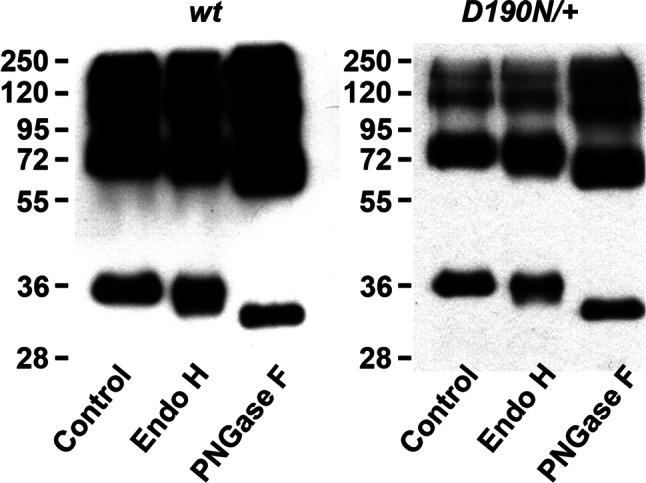
D190N, a missense mutation in rhodopsin, causes photoreceptor degeneration in patients with autosomal dominant retinitis pigmentosa (adRP). Two competing hypotheses have been developed to explain why D190N rod photoreceptors degenerate: (a) defective rhodopsin trafficking prevents proteins from correctly exiting the endoplasmic reticulum, leading to their accumulation, with deleterious effects or (b) elevated mutant rhodopsin expression and unabated signaling causes excitotoxicity. A knock-in D190N mouse model was engineered to delineate the mechanism of pathogenesis. Wild type (wt) and mutant rhodopsin appeared correctly localized in rod outer segments of D190N heterozygotes. Moreover, the rhodopsin glycosylation state in the mutants appeared similar to that in wt mice. Thus, it seems plausible that the injurious effect of the heterozygous mutation is not related to mistrafficking of the protein, but rather from constitutive rhodopsin activity and a greater propensity for chromophore isomerization even in the absence of light.
Keywords: D190N; Excitotoxicity; GPCR; Mouse model; Retina; Retinitis pigmentosa; Rhodopsin.
Figures
References
-
- Berson EL. Retinitis pigmentosa: the friedenwald lecture. Invest Opthal Vis Sci. 1993;34:1655–1676. - PubMed
MeSH terms
Substances
Grants and funding
- T32 HL007344/HL/NHLBI NIH HHS/United States
- R01EY018213/NH/NIH HHS/United States
- R01 EY018213/EY/NEI NIH HHS/United States
- R01EY016822/NH/NIH HHS/United States
- PROMETEO/2016/094/Prometeo grant
- K08 EY020530/EY/NEI NIH HHS/United States
- R01 EY026682/EY/NEI NIH HHS/United States
- F30EYE027986/NH/NIH HHS/United States
- T32GM007337/NH/NIH HHS/United States
- T32 GM007337/GM/NIGMS NIH HHS/United States
- R01EY026682/NH/NIH HHS/United States
- P30EY019007/NH/NIH HHS/United States
- K08EY020530/NH/NIH HHS/United States
- R21 AG050437/AG/NIA NIH HHS/United States
- TS080017/U.S. Department of Defense
- R01 EY016822/EY/NEI NIH HHS/United States
- R21AG050437/NH/NIH HHS/United States
- 2013103/DDCF/Doris Duke Charitable Foundation/United States
- TA-NMT-0116-0692-COLU/FFB/Foundation Fighting Blindness/United States
- R01 EY024698/EY/NEI NIH HHS/United States
- P30 EY019007/EY/NEI NIH HHS/United States
- N09G-302/New York State
- WT_/Wellcome Trust/United Kingdom
- 07-CS3/Charles E. Culpeper Partnership for Cures
- BEST/2016/030/BEST2016
- R01EY024698/NH/NIH HHS/United States
LinkOut - more resources
Full Text Sources
Research Materials
