

A Compendium of Mutational Signatures of Environmental Agents
- PMID: 30982602
- PMCID: PMC6506336
- DOI: 10.1016/j.cell.2019.03.001
A Compendium of Mutational Signatures of Environmental Agents
Abstract
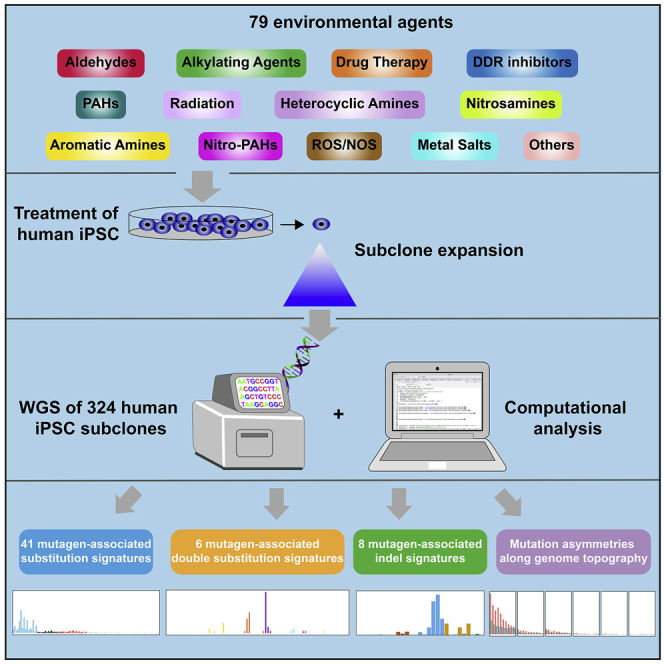
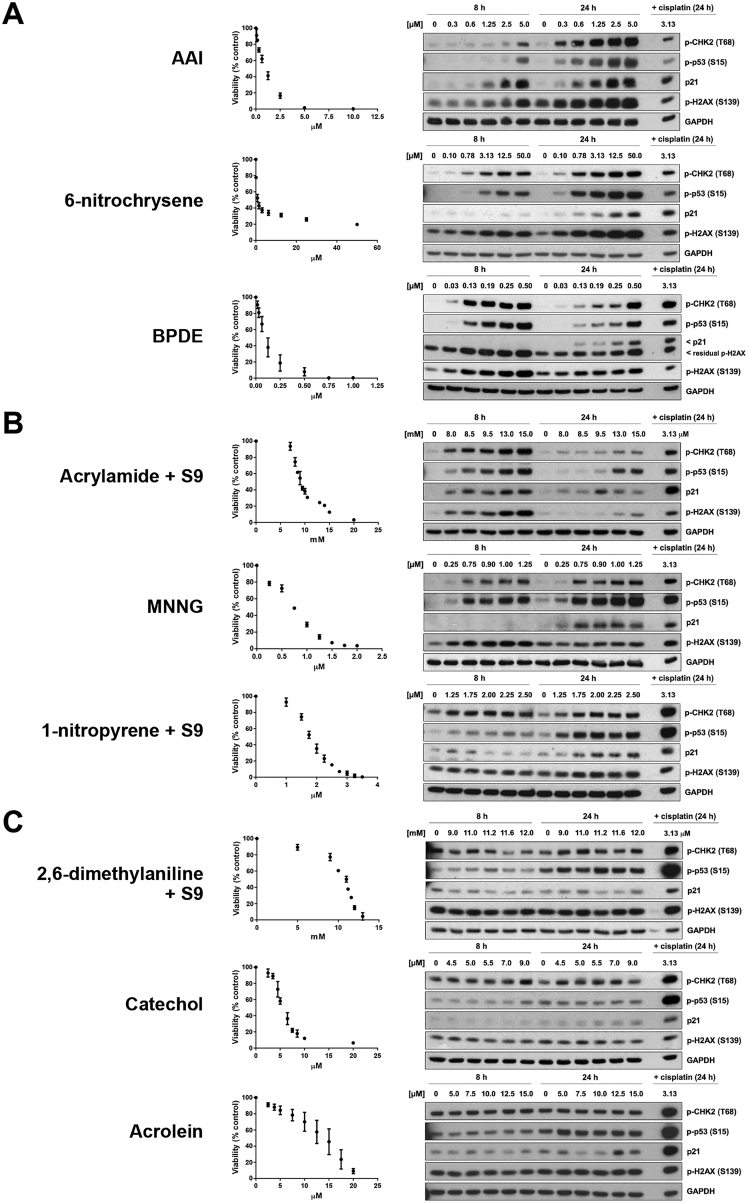
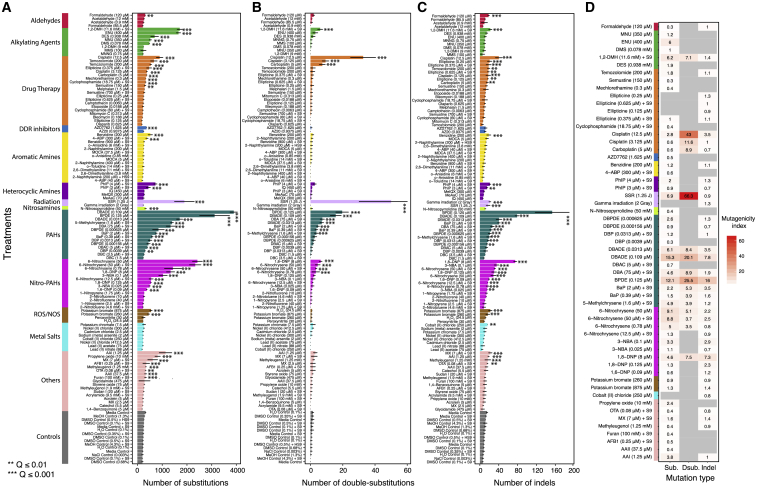
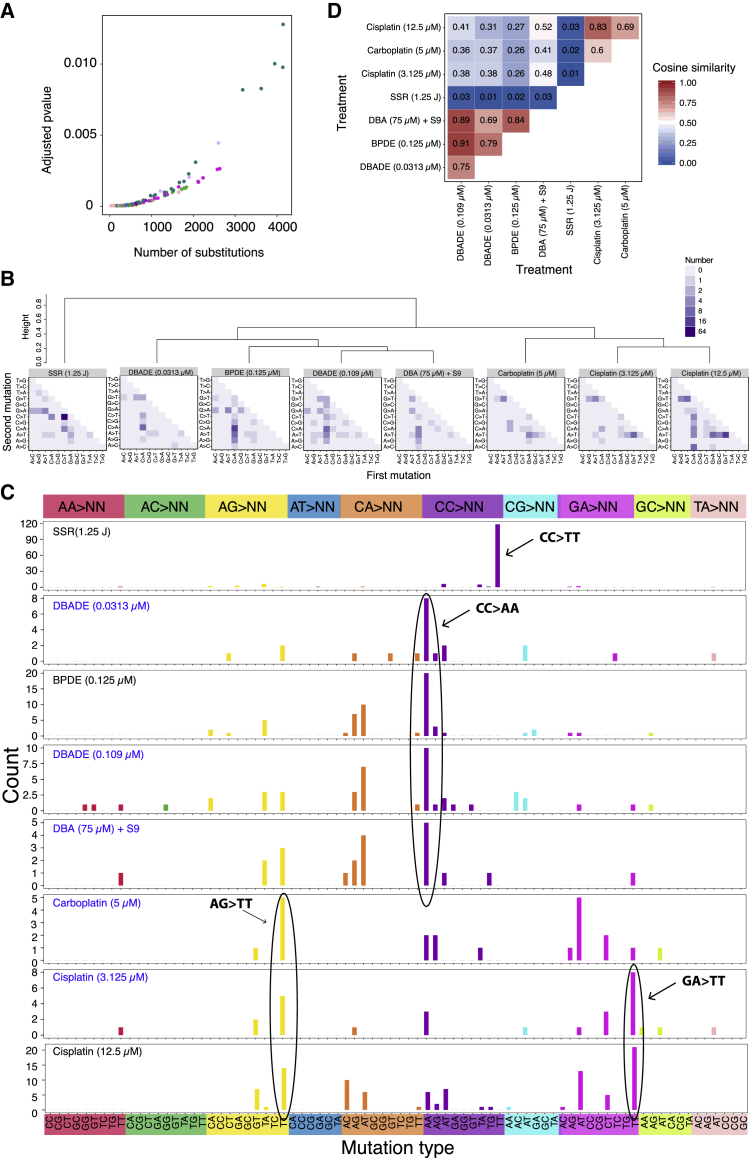
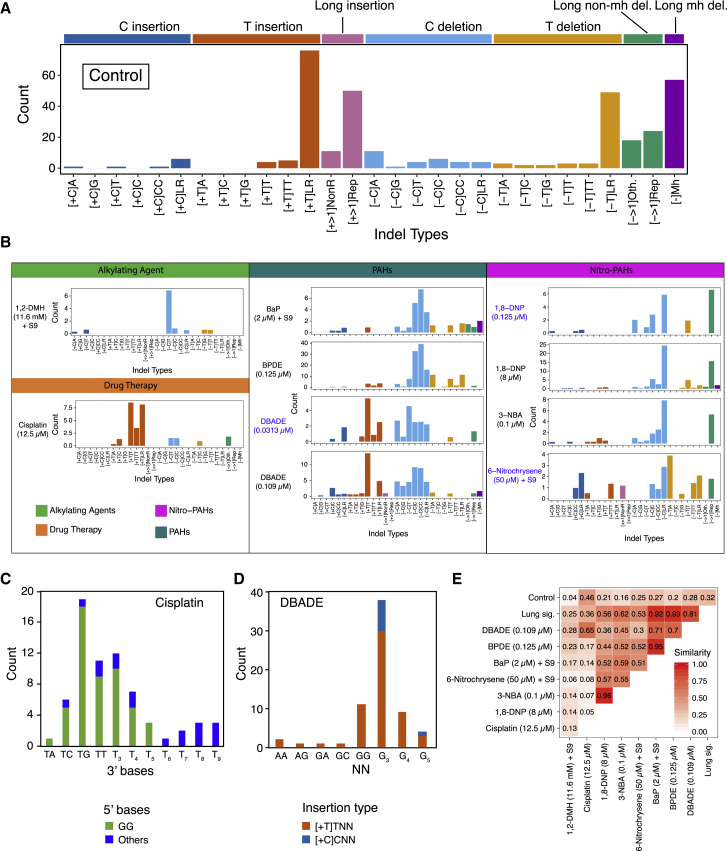
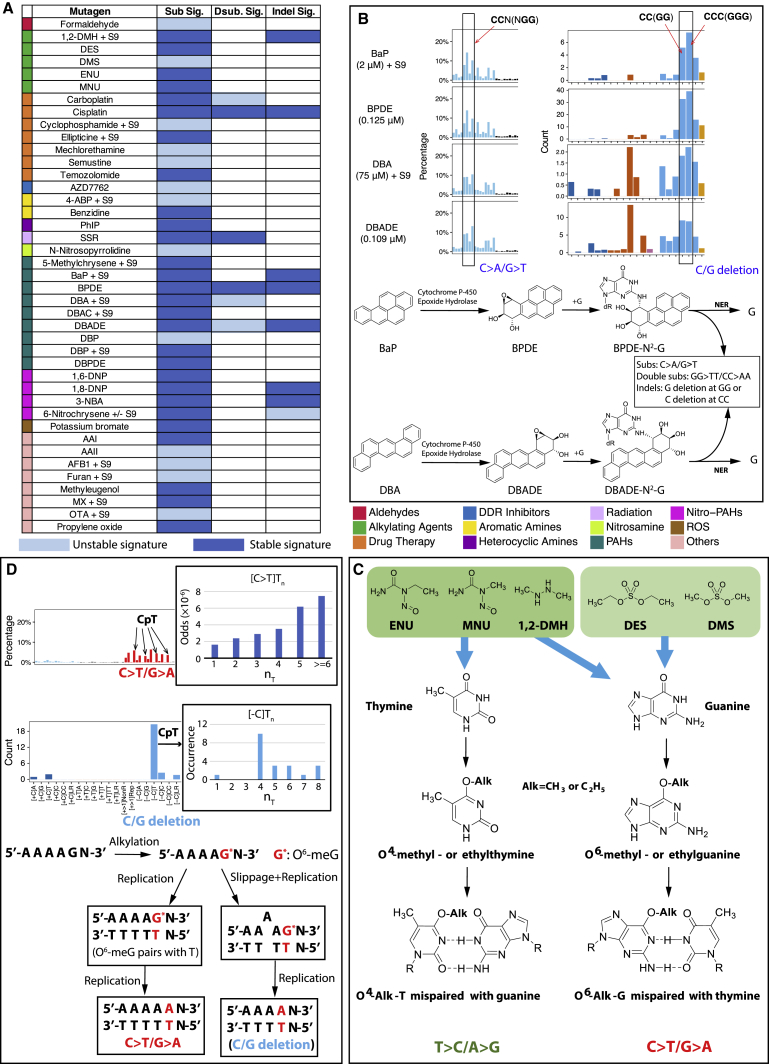
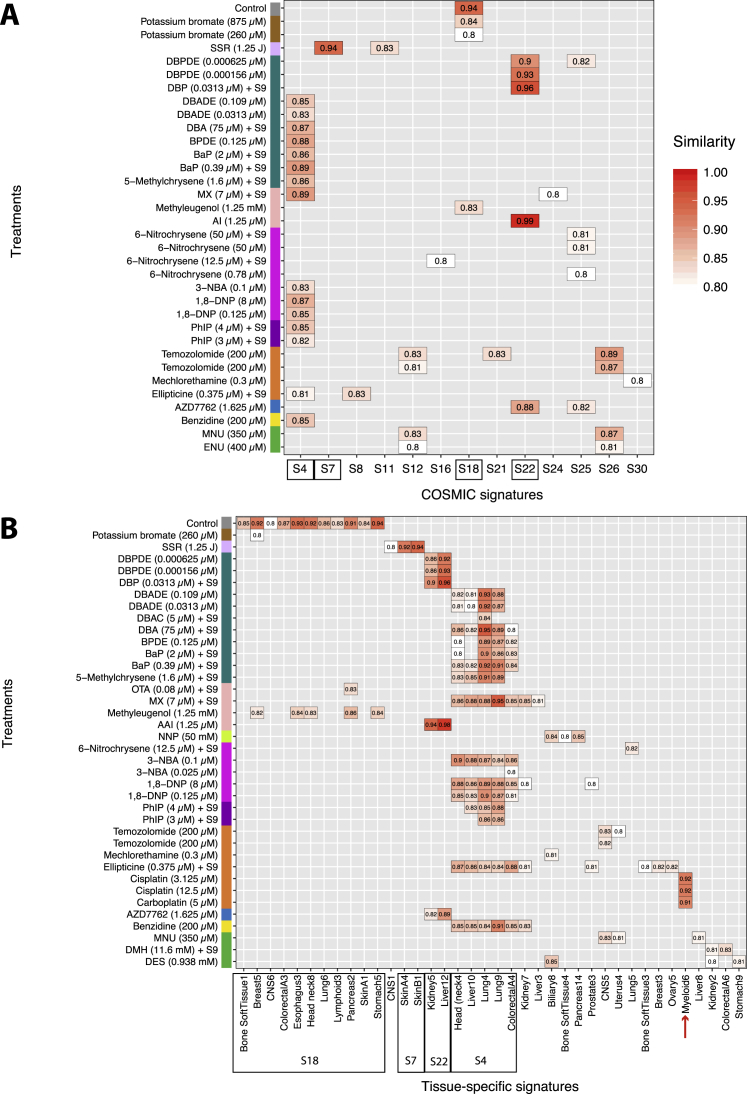
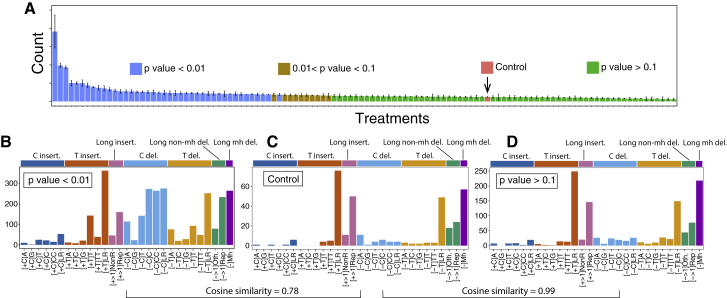
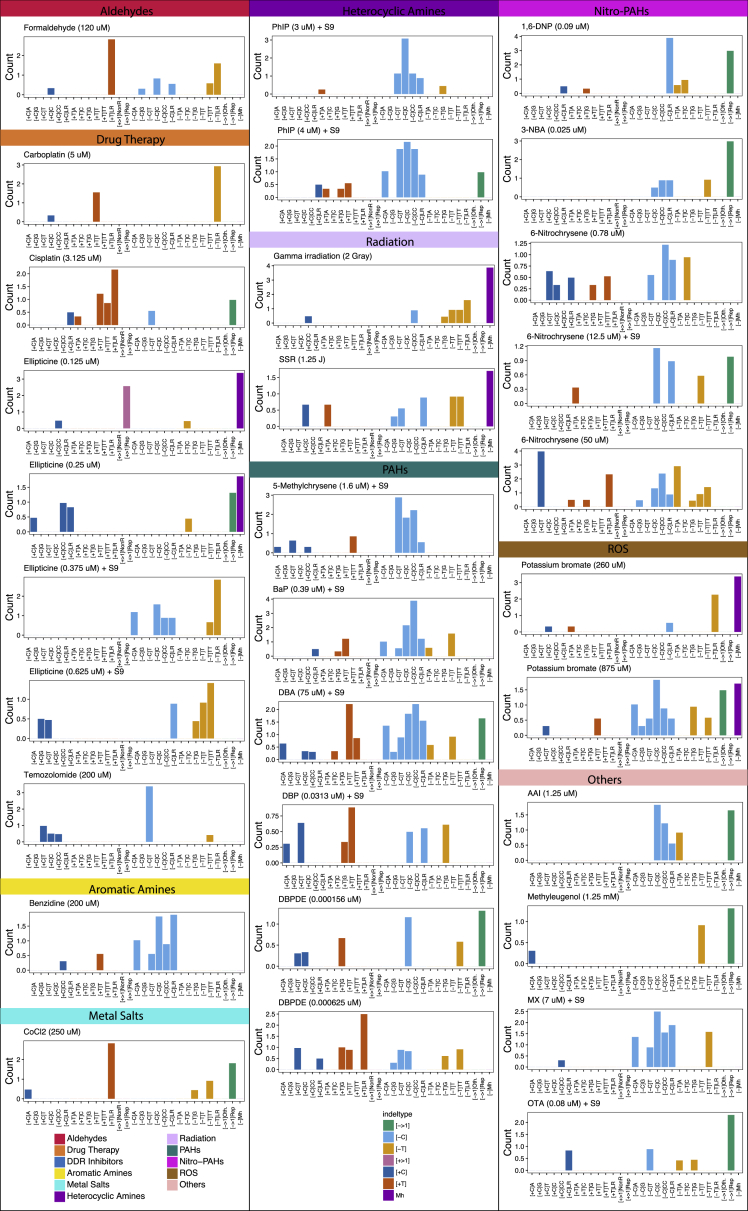
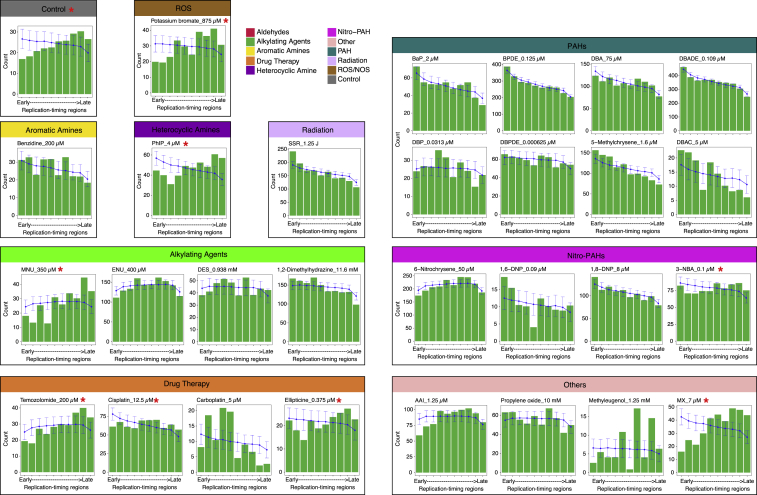
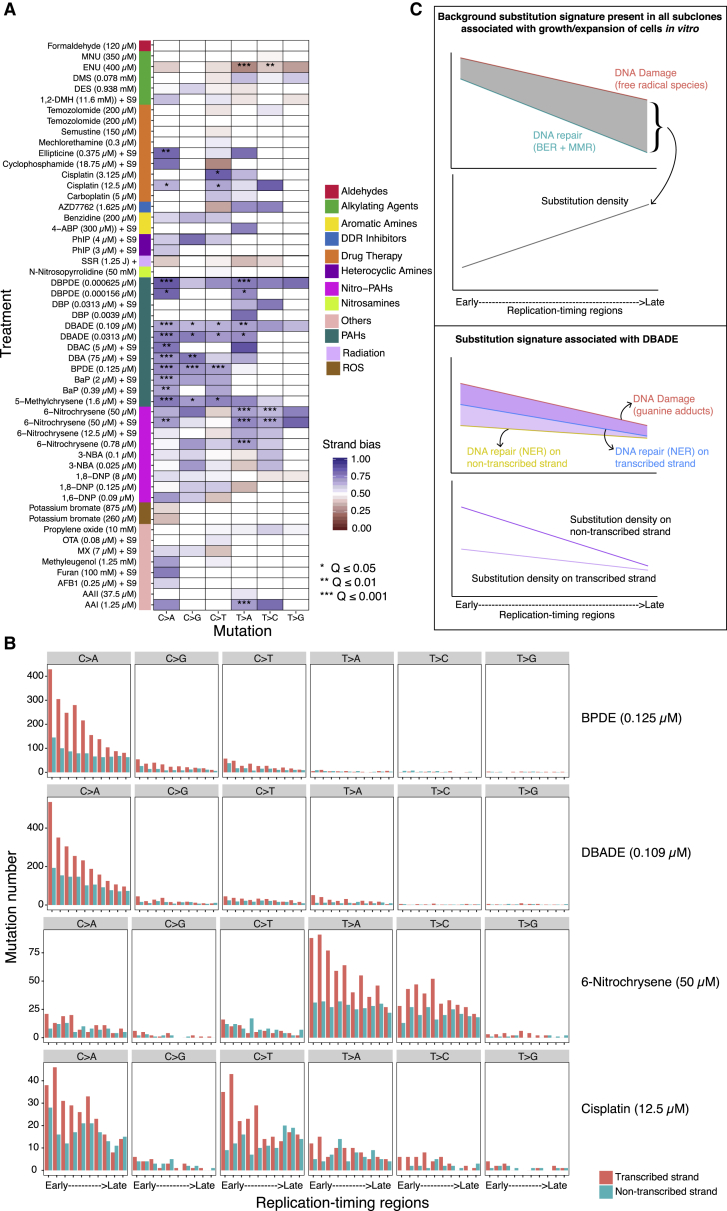
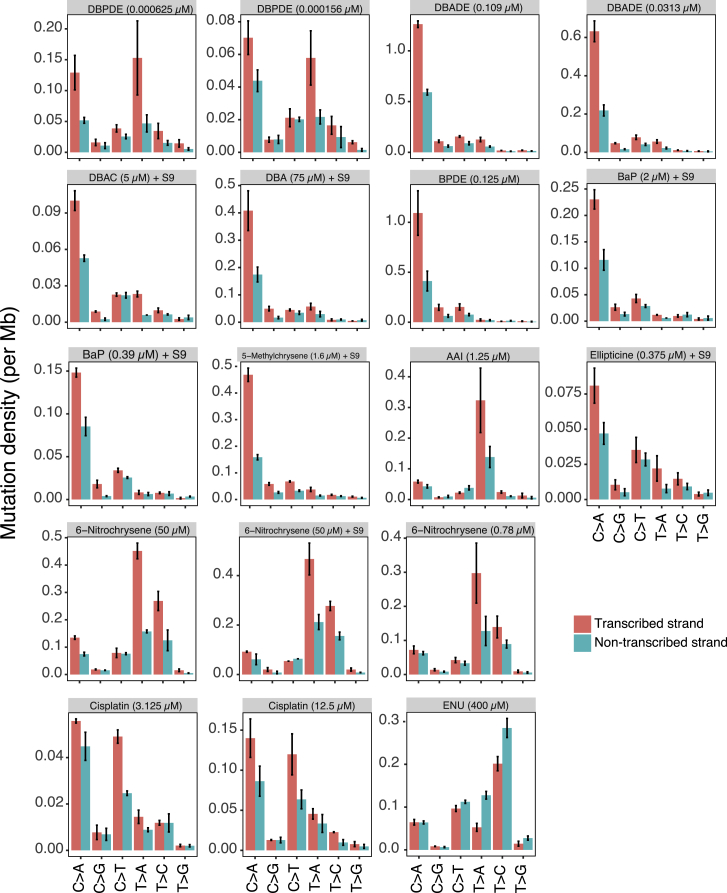
Whole-genome-sequencing (WGS) of human tumors has revealed distinct mutation patterns that hint at the causative origins of cancer. We examined mutational signatures in 324 WGS human-induced pluripotent stem cells exposed to 79 known or suspected environmental carcinogens. Forty-one yielded characteristic substitution mutational signatures. Some were similar to signatures found in human tumors. Additionally, six agents produced double-substitution signatures and eight produced indel signatures. Investigating mutation asymmetries across genome topography revealed fully functional mismatch and transcription-coupled repair pathways. DNA damage induced by environmental mutagens can be resolved by disparate repair and/or replicative pathways, resulting in an assortment of signature outcomes even for a single agent. This compendium of experimentally induced mutational signatures permits further exploration of roles of environmental agents in cancer etiology and underscores how human stem cell DNA is directly vulnerable to environmental agents. VIDEO ABSTRACT.
Keywords: DNA damage; Mutational signatures; carcinogens; environmental mutagens; mutagenesis.
Copyright © 2019 The Author(s). Published by Elsevier Inc. All rights reserved.
Figures



References
-
- Alexandrov L.B., Nik-Zainal S., Wedge D.C., Aparicio S.A.J.R., Behjati S., Biankin A.V., Bignell G.R., Bolli N., Borg A., Børresen-Dale A.-L., Australian Pancreatic Cancer Genome Initiative. ICGC Breast Cancer Consortium. ICGC MMML-Seq Consortium. ICGC PedBrain Signatures of mutational processes in human cancer. Nature. 2013;500:415–421. - PMC - PubMed
-
- Arlt V.M., Glatt H., Muckel E., Pabel U., Sorg B.L., Schmeiser H.H., Phillips D.H. Metabolic activation of the environmental contaminant 3-nitrobenzanthrone by human acetyltransferases and sulfotransferase. Carcinogenesis. 2002;23:1937–1945. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
