

Age-dependent accumulation of oligomeric SNCA/α-synuclein from impaired degradation in mutant LRRK2 knockin mouse model of Parkinson disease: role for therapeutic activation of chaperone-mediated autophagy (CMA)
- PMID: 30983487
- PMCID: PMC6984454
- DOI: 10.1080/15548627.2019.1603545
Age-dependent accumulation of oligomeric SNCA/α-synuclein from impaired degradation in mutant LRRK2 knockin mouse model of Parkinson disease: role for therapeutic activation of chaperone-mediated autophagy (CMA)
Abstract
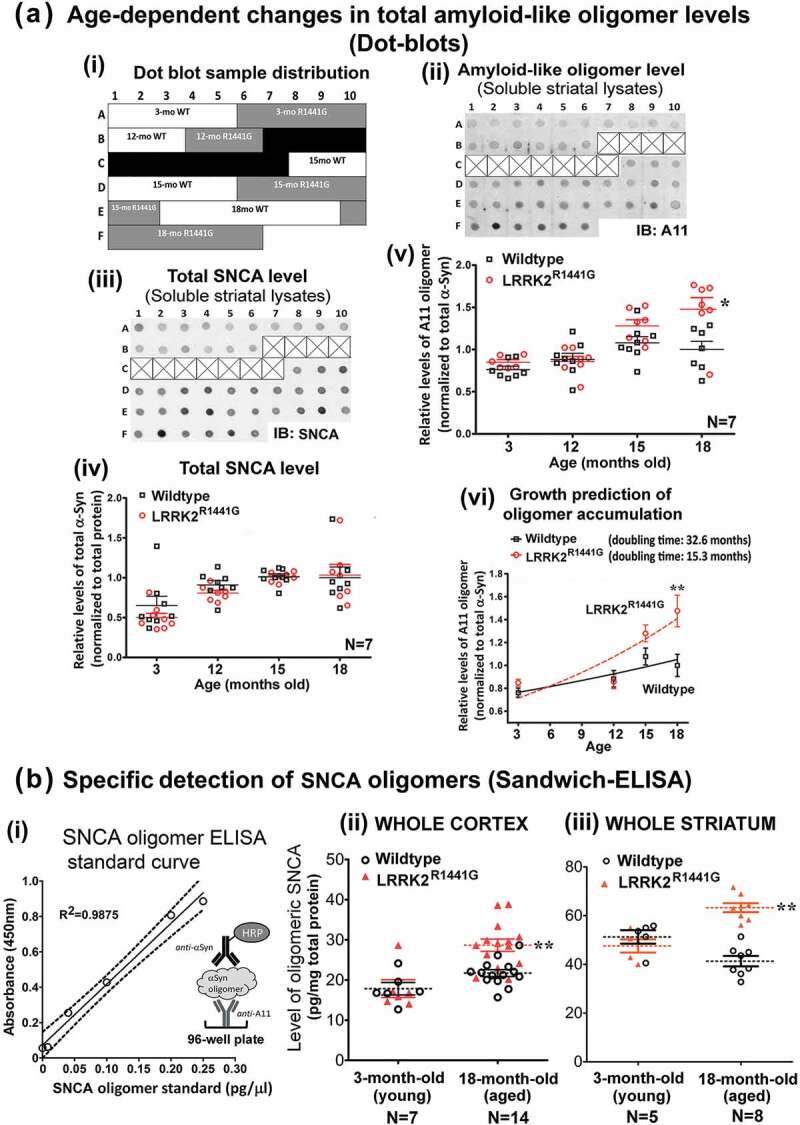
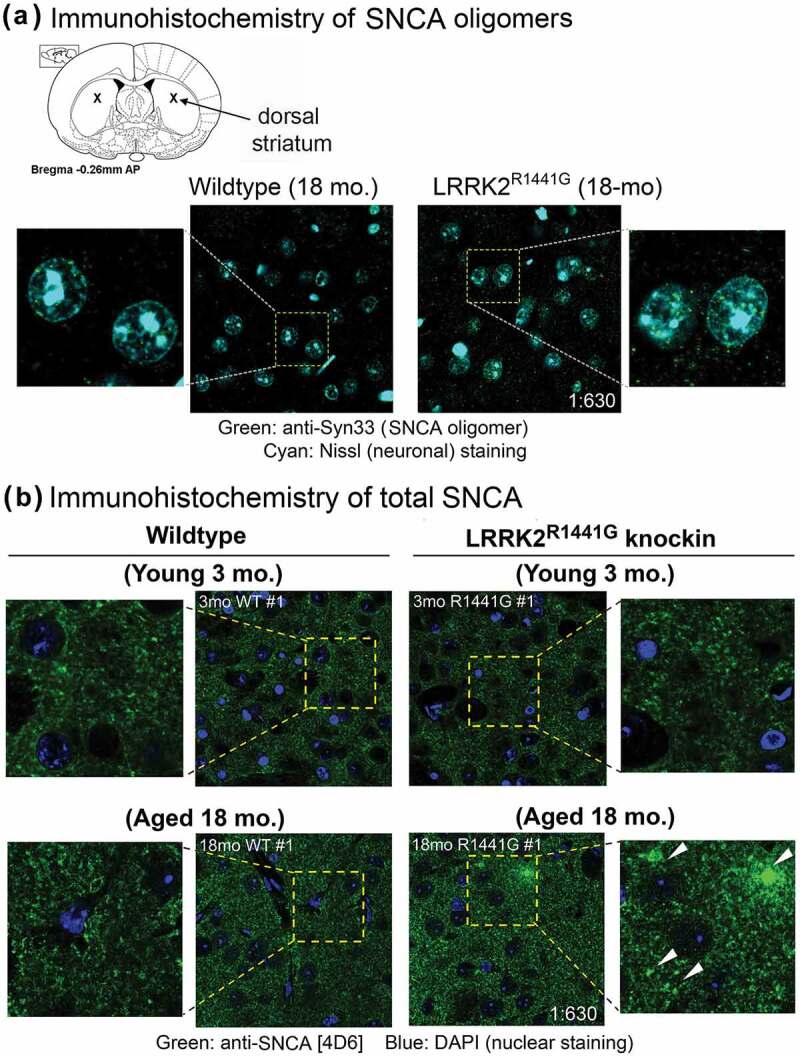
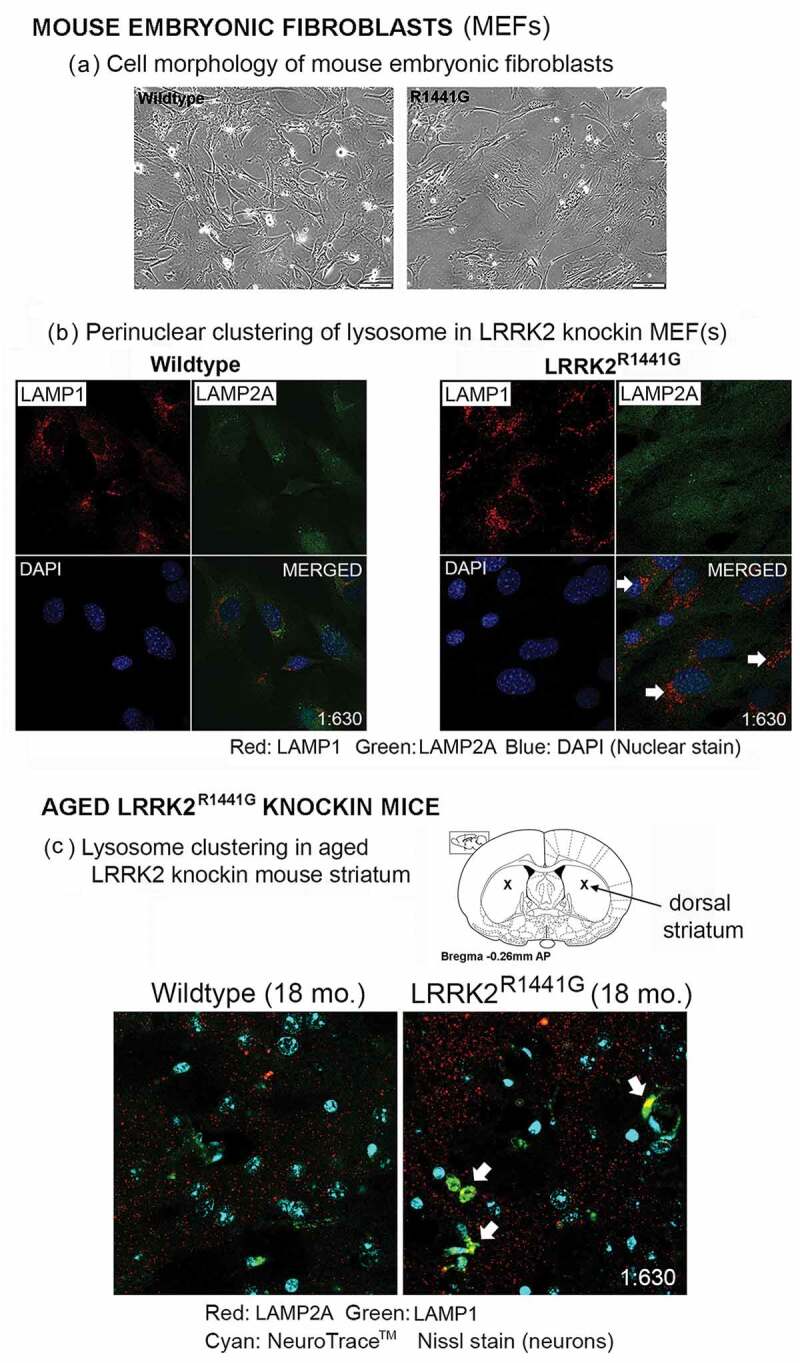
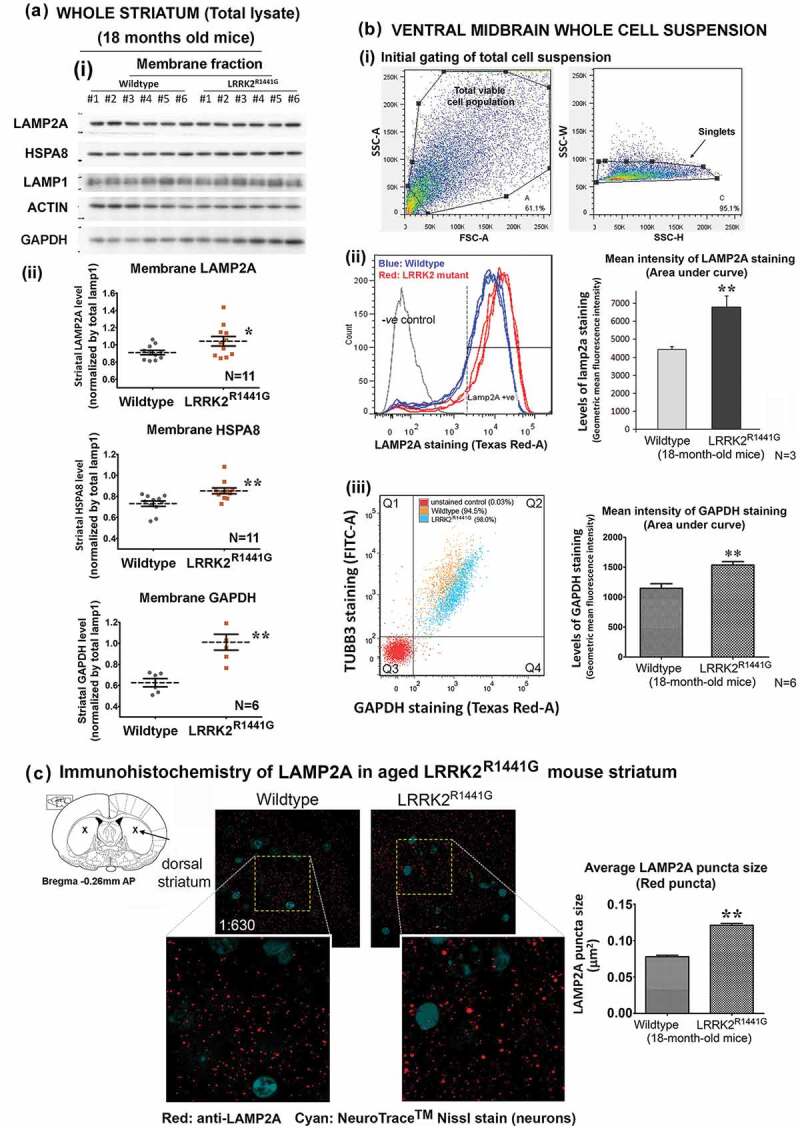
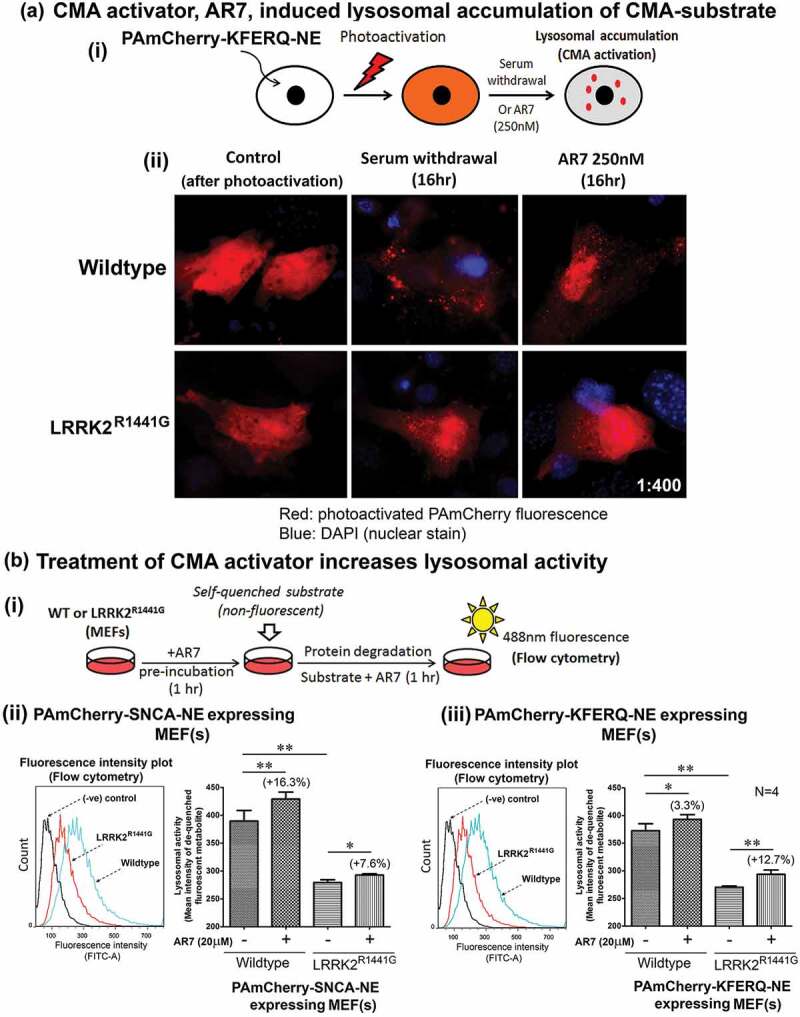
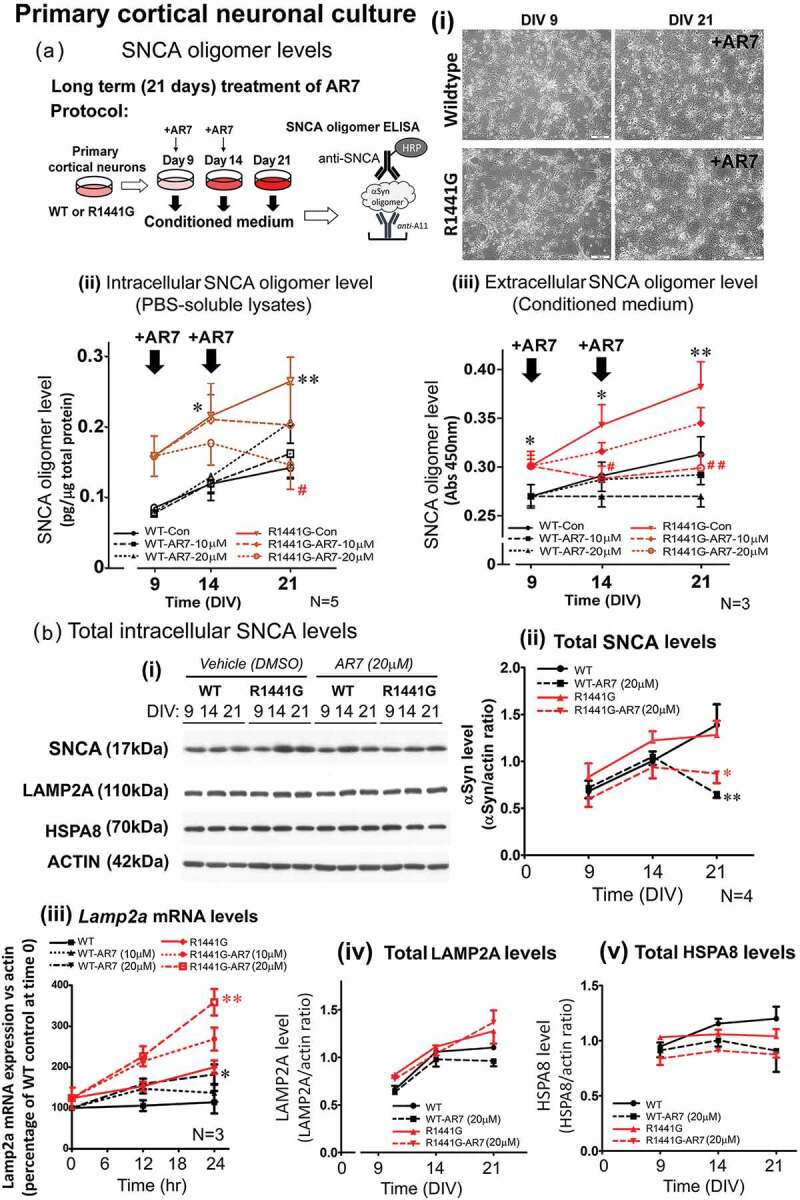
Parkinson disease (PD) is an age-related neurodegenerative disorder associated with misfolded SNCA/α-synuclein accumulation in brain. Impaired catabolism of SNCA potentiates formation of its toxic oligomers. LRRK2 (leucine-rich repeat kinase-2) mutations predispose to familial and sporadic PD. Mutant LRRK2 perturbs chaperone-mediated-autophagy (CMA) to degrade SNCA. We showed greater age-dependent accumulation of oligomeric SNCA in striatum and cortex of aged LRRK2R1441G knockin (KI) mice, compared to age-matched wildtype (WT) by 53% and 31%, respectively. Lysosomal clustering and accumulation of CMA-specific LAMP2A and HSPA8/HSC70 proteins were observed in aged mutant striatum along with increased GAPDH (CMA substrate) by immunohistochemistry of dorsal striatum and flow cytometry of ventral midbrain cells. Using our new reporter protein clearance assay, mutant mouse embryonic fibroblasts (MEFs) expressing either SNCA or CMA recognition 'KFERQ'-like motif conjugated with photoactivated-PAmCherry showed slower cellular clearance compared to WT by 28% and 34%, respectively. However, such difference was not observed after the 'KFERQ'-motif was mutated. LRRK2 mutant MEFs exhibited lower lysosomal degradation than WT indicating lysosomal dysfunction. LAMP2A-knockdown reduced total lysosomal activity and clearance of 'KFERQ'-substrate in WT but not in mutant MEFs, indicating impaired CMA in the latter. A CMA-specific activator, AR7, induced neuronal LAMP2A transcription and lysosomal activity in MEFs. AR7 also attenuated the progressive accumulation of both intracellular and extracellular SNCA oligomers in prolonged cultures of mutant cortical neurons (DIV21), indicating that oligomer accumulation can be suppressed by CMA activation. Activation of autophagic pathways to reduce aged-related accumulation of pathogenic SNCA oligomers is a viable disease-modifying therapeutic strategy for PD.Abbreviations: 3-MA: 3-methyladenine; AR7: 7-chloro-3-(4-methylphenyl)-2H-1,4-benzoxazine; CMA: chaperone-mediated autophagy; CQ: chloroquine; CSF: cerebrospinal fluid; DDM: n-dodecyl β-D-maltoside; DIV: days in vitro; ELISA: enzyme-linked immunosorbent assay; FACS: fluorescence-activated cell sorting; GAPDH: glyceraldehyde-3-phosphate dehydrogenase; GWAS: genome-wide association studies; HSPA8/HSC70: heat shock protein 8; KFERQ: CMA recognition pentapeptide; KI: knockin; LAMP1: lysosomal-associated membrane protein 1; LAMP2A: lysosomal-associated membrane protein 2A; LDH: lactate dehydrogenase; LRRK2: leucine-rich repeat kinase 2; MEF: mouse embryonic fibroblast; NDUFS4: NADH:ubiquinone oxidoreductase core subunit S4; NE: novel epitope; PD: Parkinson disease; RARA/RARα: retinoic acid receptor, alpha; SNCA: synuclein, alpha; TUBB3/TUJ1: tubulin, beta 3 class III; WT: wild-type.
Keywords: Alpha-synuclein; LRRK2; Parkinson disease; aging; chaperone-mediated autophagy; knockin mouse model; oligomers; protein aggregation; protein degradation; therapeutic strategy.
Figures
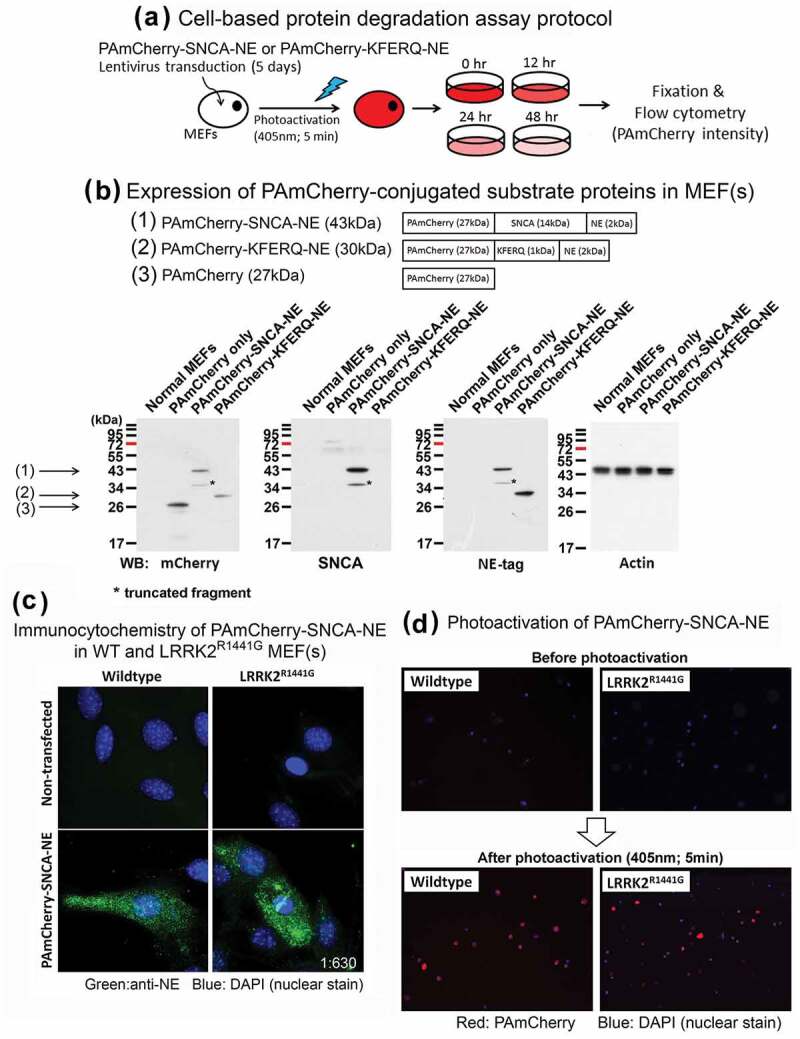
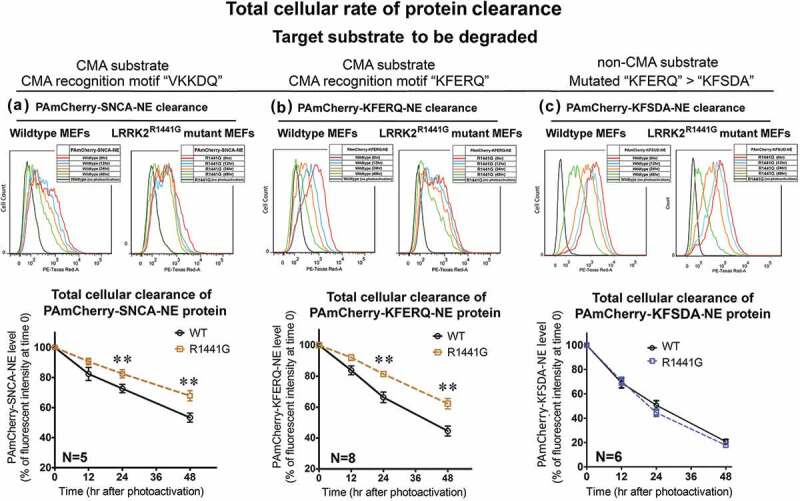
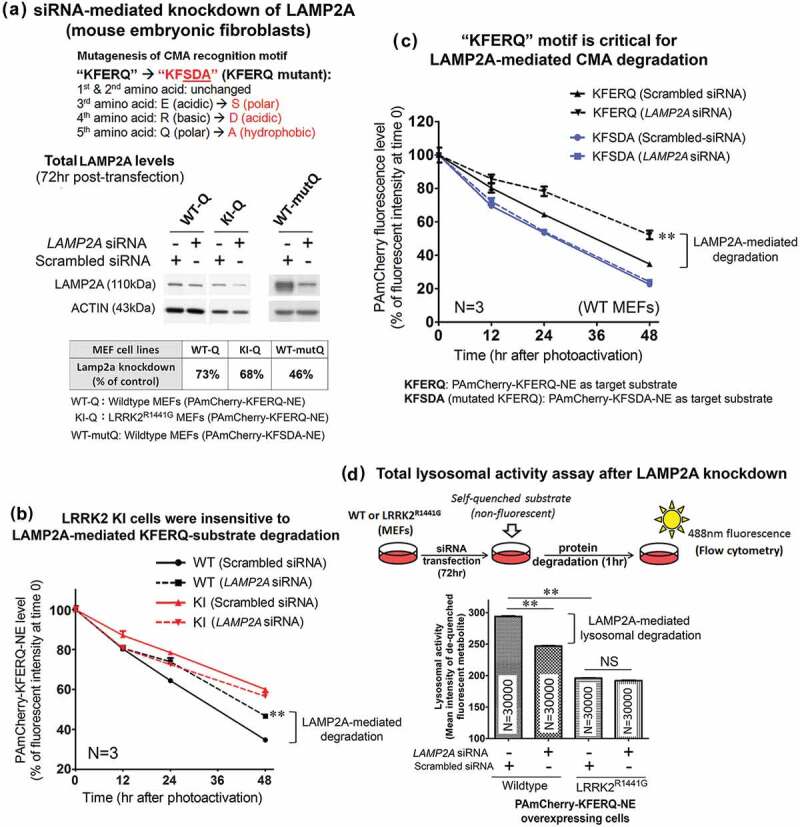
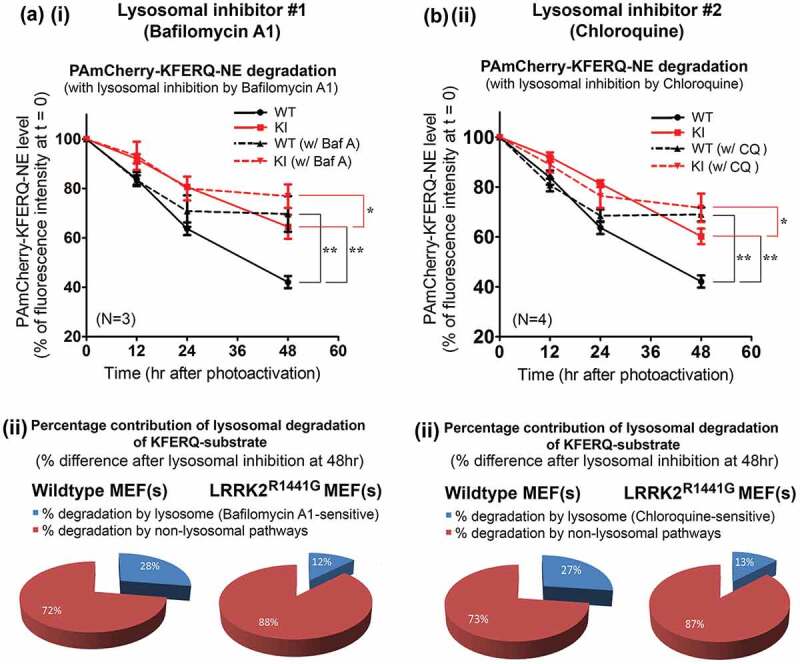
References
-
- de Lau LM, Breteler MM.. Epidemiology of Parkinson’s disease. Lancet Neurol. 2006;5(6):525–535. - PubMed
-
- Lesage S, Brice A.. Parkinson‘s disease: from monogenic forms to genetic susceptibility factors. Hum Mol Genet. 2009;18(R1):R48–R59. - PubMed
-
- Verstraeten A, Theuns J, Van Broeckhoven C. Progress in unraveling the genetic etiology of Parkinson disease in a genomic era. Trends Genet. 2015;31(3):140–149. - PubMed
-
- Satake W, Nakabayashi Y, Mizuta I, et al. Genome-wide association study identifies common variants at four loci as genetic risk factors for Parkinson’s disease. Nat Genet. 2009;41:1303–1307. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous