

Uncovering Missing Heritability in Rare Diseases
- PMID: 30987386
- PMCID: PMC6523881
- DOI: 10.3390/genes10040275
Uncovering Missing Heritability in Rare Diseases
Abstract
The problem of 'missing heritability' affects both common and rare diseases hindering: discovery, diagnosis, and patient care. The 'missing heritability' concept has been mainly associated with common and complex diseases where promising modern technological advances, like genome-wide association studies (GWAS), were unable to uncover the complete genetic mechanism of the disease/trait. Although rare diseases (RDs) have low prevalence individually, collectively they are common. Furthermore, multi-level genetic and phenotypic complexity when combined with the individual rarity of these conditions poses an important challenge in the quest to identify causative genetic changes in RD patients. In recent years, high throughput sequencing has accelerated discovery and diagnosis in RDs. However, despite the several-fold increase (from ~10% using traditional to ~40% using genome-wide genetic testing) in finding genetic causes of these diseases in RD patients, as is the case in common diseases-the majority of RDs are also facing the 'missing heritability' problem. This review outlines the key role of high throughput sequencing in uncovering genetics behind RDs, with a particular focus on genome sequencing. We review current advances and challenges of sequencing technologies, bioinformatics approaches, and resources.
Keywords: bioinformatics; genome sequencing; long/short read sequencing; missing heritability; rare disease; variant annotation; variant detection; variation databases.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
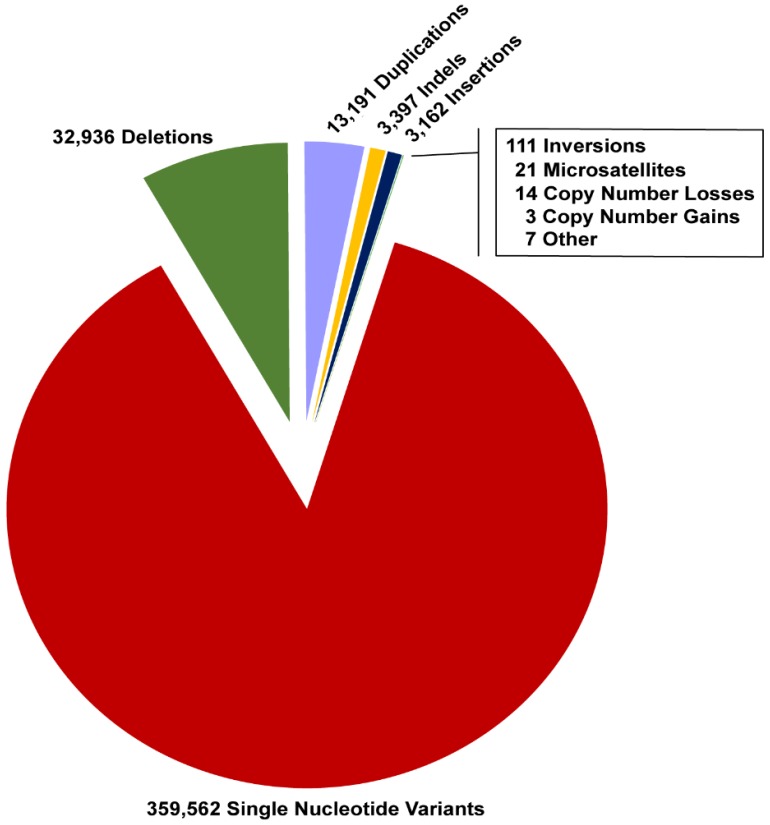

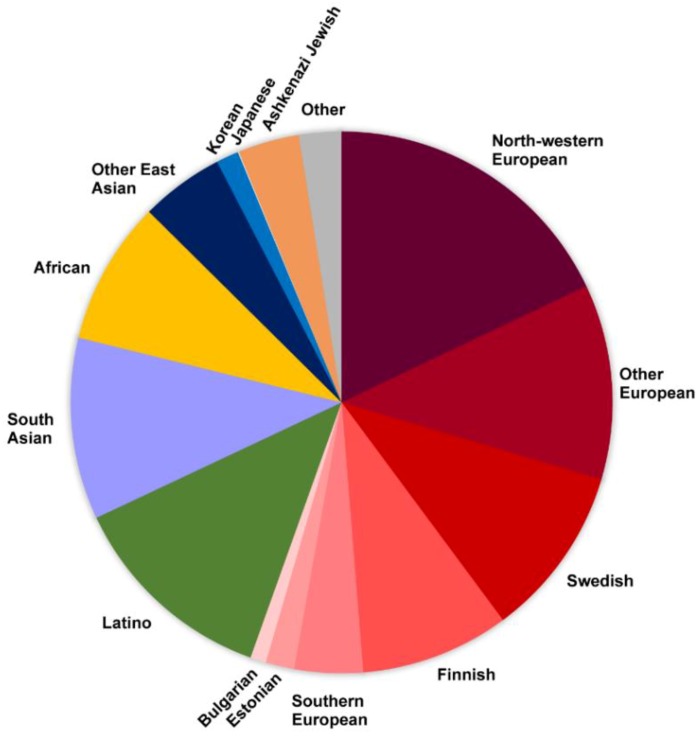
References
-
- Turkheimer E. Still missing. Res. Hum. Dev. 2011;8:227–241. doi: 10.1080/15427609.2011.625321. - DOI
-
- Montserrat Moliner A., Waligóra J. The European Union policy in the field of rare diseases. Public Health Genomics. 2013;16:268–277. - PubMed
-
- Orphanet. [(accessed on 6 January 2019)]; Available online: https://www.orpha.net/consor/cgi-bin/index.php.
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous
