

Growth Factor Independence 1B-Mediated Transcriptional Repression and Lineage Allocation Require Lysine-Specific Demethylase 1-Dependent Recruitment of the BHC Complex
- PMID: 30988160
- PMCID: PMC6580704
- DOI: 10.1128/MCB.00020-19
Growth Factor Independence 1B-Mediated Transcriptional Repression and Lineage Allocation Require Lysine-Specific Demethylase 1-Dependent Recruitment of the BHC Complex
Abstract
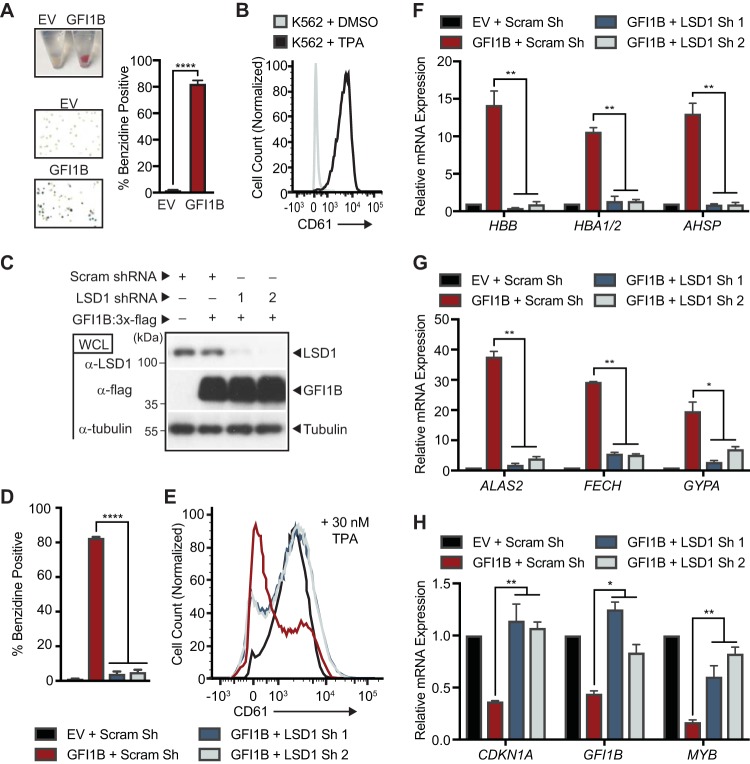
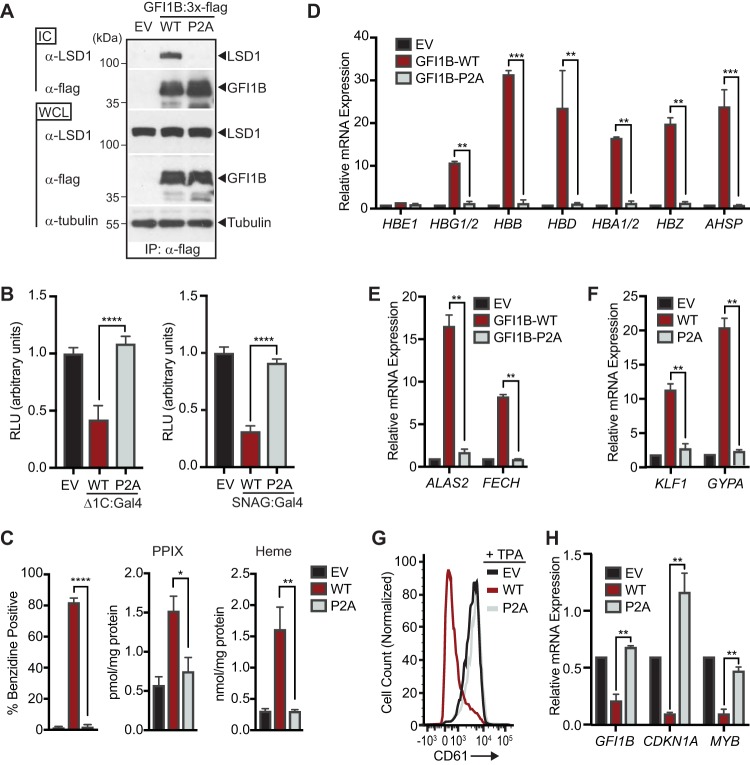
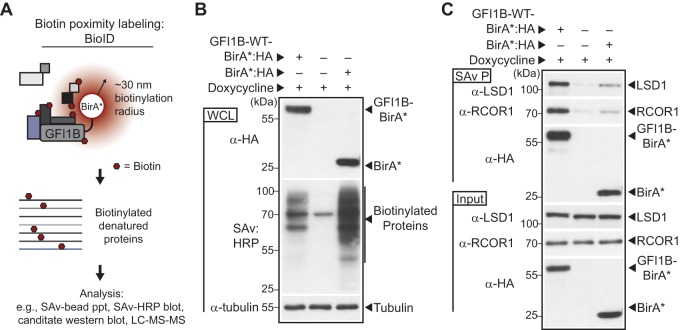
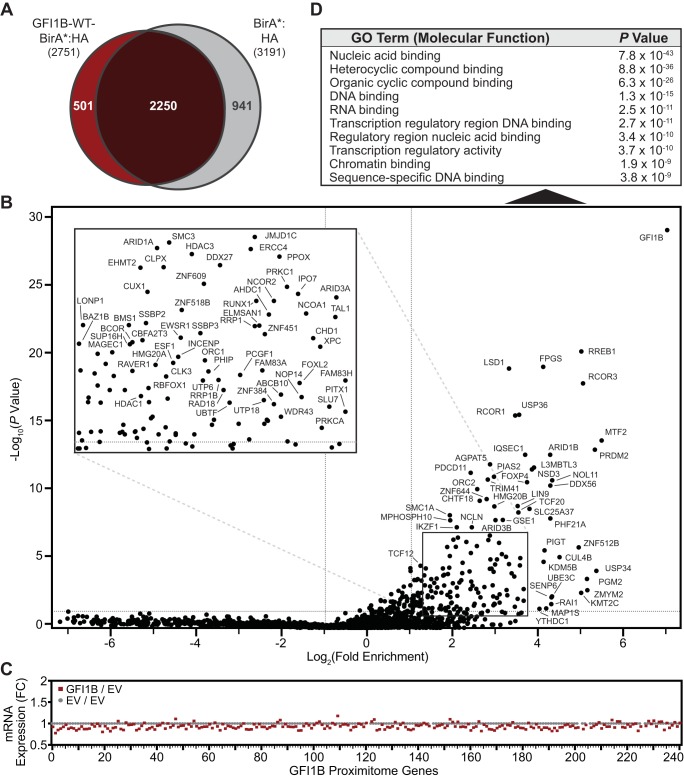
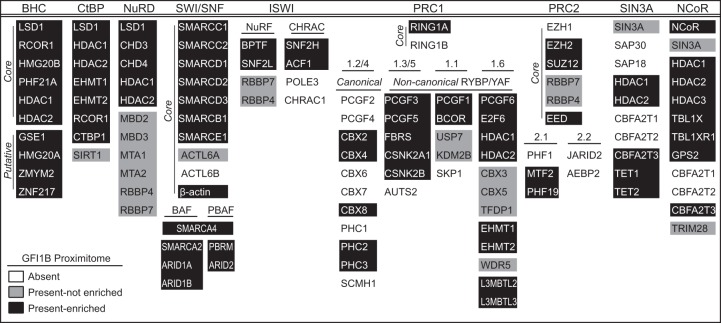
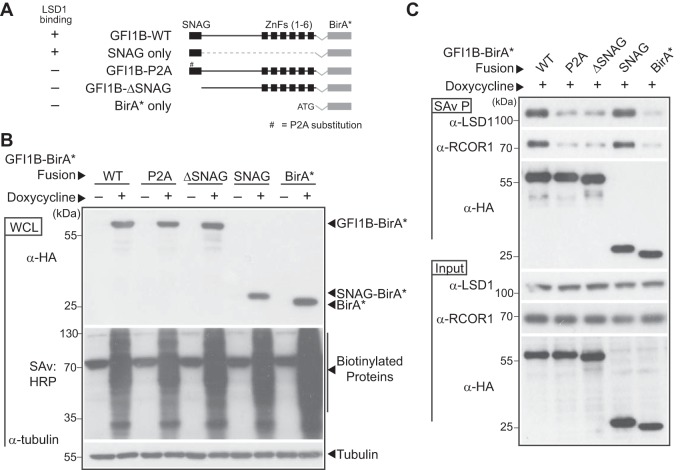
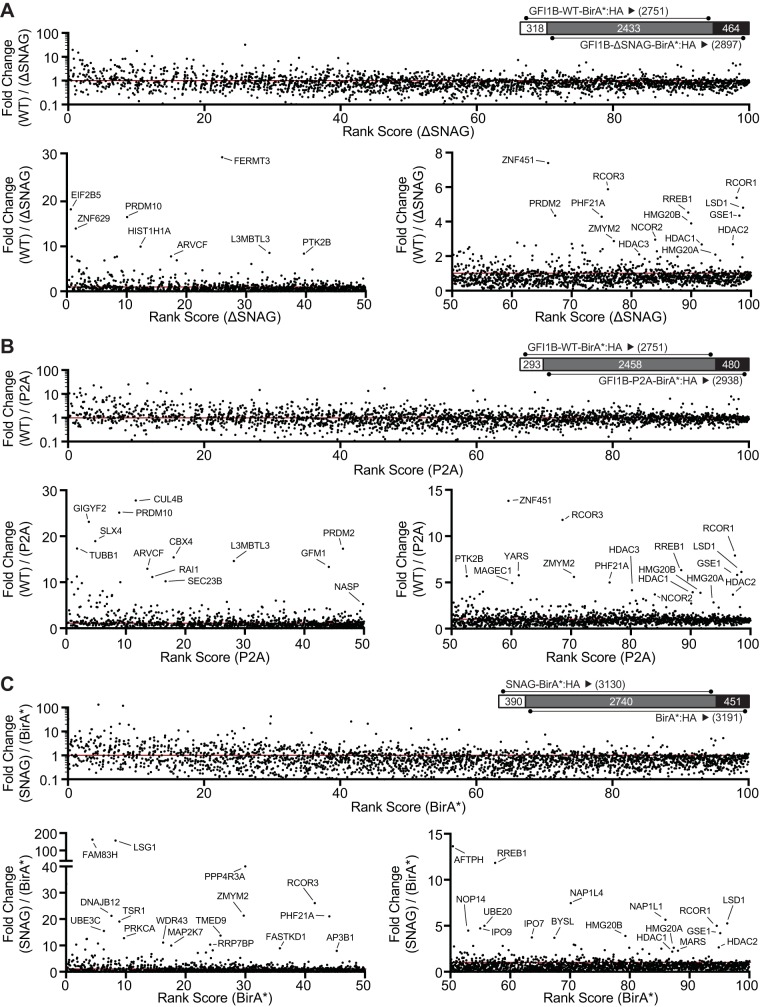
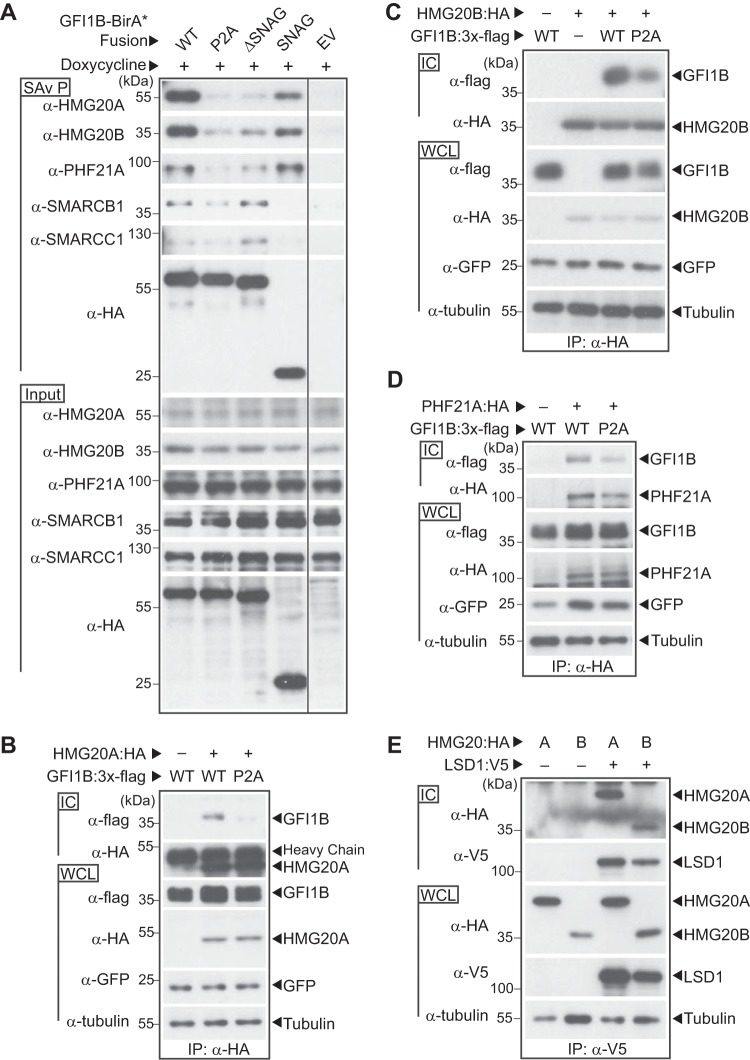
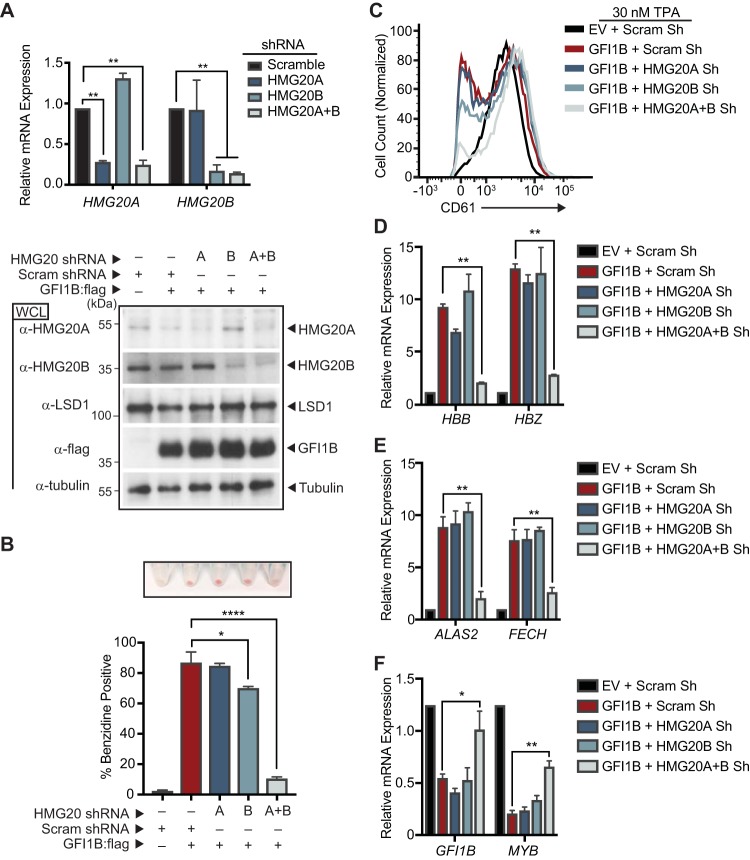
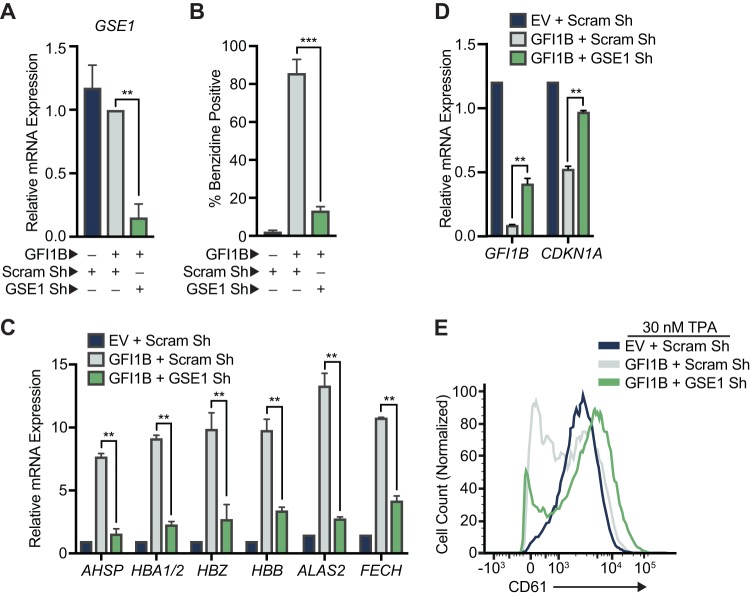
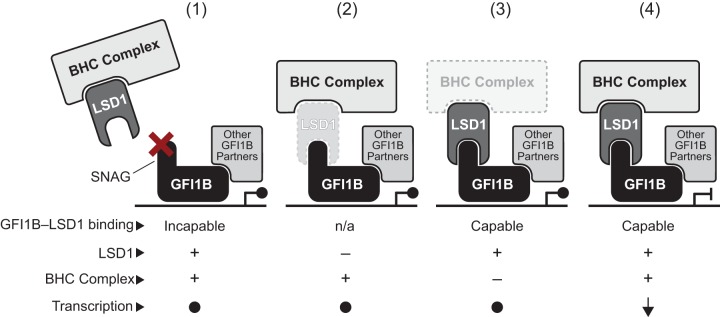
Growth factor independence 1B (GFI1B) coordinates assembly of transcriptional repressor complexes comprised of corepressors and histone-modifying enzymes to control gene expression programs governing lineage allocation in hematopoiesis. Enforced expression of GFI1B in K562 erythroleukemia cells favors erythroid over megakaryocytic differentiation, providing a platform to define molecular determinants of binary fate decisions triggered by GFI1B. We deployed proteome-wide proximity labeling to identify factors whose inclusion in GFI1B complexes depends upon GFI1B's obligate effector, lysine-specific demethylase 1 (LSD1). We show that GFI1B preferentially recruits core and putative elements of the BRAF-histone deacetylase (HDAC) (BHC) chromatin-remodeling complex (LSD1, RCOR1, HMG20A, HMG20B, HDAC1, HDAC2, PHF21A, GSE1, ZMYM2, and ZNF217) in an LSD1-dependent manner to control acquisition of erythroid traits by K562 cells. Among these elements, depletion of both HMG20A and HMG20B or of GSE1 blocks GFI1B-mediated erythroid differentiation, phenocopying impaired differentiation brought on by LSD1 depletion or disruption of GFI1B-LSD1 binding. These findings demonstrate the central role of the GFI1B-LSD1 interaction as a determinant of BHC complex recruitment to enable cell fate decisions driven by GFI1B.
Keywords: BHC complex; Erythropoiesis; GFI1B; GSE1; HMG20A; HMG20B; LSD1; SNAG domain; hematopoiesis; transcriptional repression.
Copyright © 2019 American Society for Microbiology.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials
Miscellaneous