

Targeting dePARylation selectively suppresses DNA repair-defective and PARP inhibitor-resistant malignancies
- PMID: 30989114
- PMCID: PMC6457938
- DOI: 10.1126/sciadv.aav4340
Targeting dePARylation selectively suppresses DNA repair-defective and PARP inhibitor-resistant malignancies
Abstract
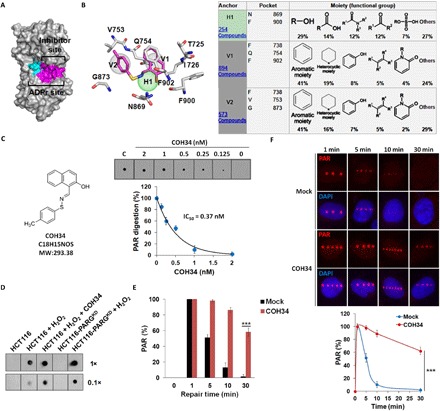
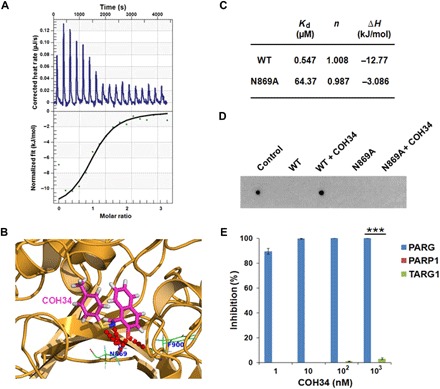
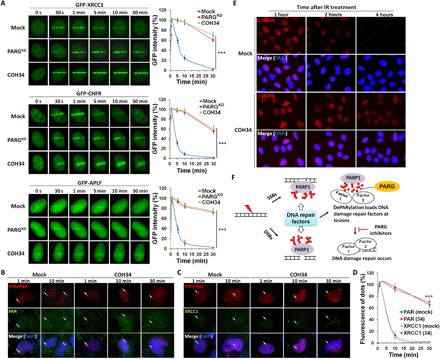
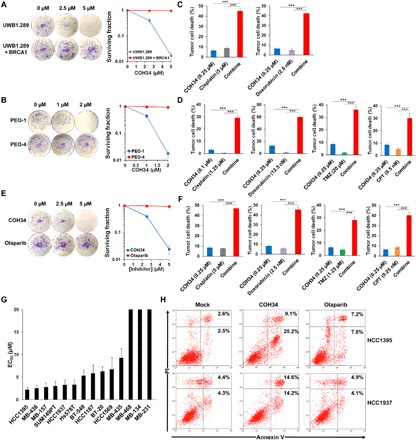
While poly(ADP-ribosyl)ation (PARylation) plays an important role in DNA repair, the role of dePARylation in DNA repair remains elusive. Here, we report that a novel small molecule identified from the NCI database, COH34, specifically inhibits poly(ADP-ribose) glycohydrolase (PARG), the major dePARylation enzyme, with nanomolar potency in vitro and in vivo. COH34 binds to the catalytic domain of PARG, thereby prolonging PARylation at DNA lesions and trapping DNA repair factors. This compound induces lethality in cancer cells with DNA repair defects and exhibits antitumor activity in xenograft mouse cancer models. Moreover, COH34 can sensitize tumor cells with DNA repair defects to other DNA-damaging agents, such as topoisomerase I inhibitors and DNA-alkylating agents, which are widely used in cancer chemotherapy. Notably, COH34 also efficiently kills PARP inhibitor-resistant cancer cells. Together, our study reveals the molecular mechanism of PARG in DNA repair and provides an effective strategy for future cancer therapies.
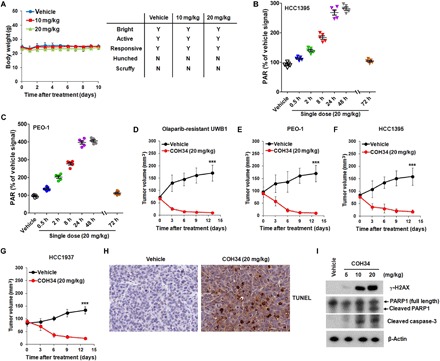
Figures
Similar articles
-
Targeting dePARylation in cancer therapy.DNA Repair (Amst). 2025 Apr;148:103824. doi: 10.1016/j.dnarep.2025.103824. Epub 2025 Mar 5. DNA Repair (Amst). 2025. PMID: 40056493 Review.
-
PARG Inhibitors and Functional PARG Inhibition Models.Curr Protein Pept Sci. 2016;17(7):641-653. doi: 10.2174/1389203717666160419145130. Curr Protein Pept Sci. 2016. PMID: 27817742
-
Targeting poly(ADP-ribose) glycohydrolase to draw apoptosis codes in cancer.Biochem Pharmacol. 2019 Sep;167:163-172. doi: 10.1016/j.bcp.2019.06.004. Epub 2019 Jun 6. Biochem Pharmacol. 2019. PMID: 31176615 Review.
-
DePARylation is critical for S phase progression and cell survival.Elife. 2024 Apr 5;12:RP89303. doi: 10.7554/eLife.89303. Elife. 2024. PMID: 38578205 Free PMC article.
-
Targeting dePARylation for cancer therapy.Cell Biosci. 2020 Jan 29;10:7. doi: 10.1186/s13578-020-0375-y. eCollection 2020. Cell Biosci. 2020. PMID: 32010441 Free PMC article. Review.
Cited by
-
PARG-deficient tumor cells have an increased dependence on EXO1/FEN1-mediated DNA repair.EMBO J. 2024 Mar;43(6):1015-1042. doi: 10.1038/s44318-024-00043-2. Epub 2024 Feb 15. EMBO J. 2024. PMID: 38360994 Free PMC article.
-
A platform to deliver single and bi-specific Cas9/guide RNA to perturb genes in vitro and in vivo.Mol Ther. 2024 Oct 2;32(10):3629-3649. doi: 10.1016/j.ymthe.2024.07.025. Epub 2024 Jul 31. Mol Ther. 2024. PMID: 39091030 Free PMC article.
-
PARG is essential for Polθ-mediated DNA end-joining by removing repressive poly-ADP-ribose marks.Nat Commun. 2024 Jul 11;15(1):5822. doi: 10.1038/s41467-024-50158-7. Nat Commun. 2024. PMID: 38987289 Free PMC article.
-
PARP inhibitor resistance in breast and gynecological cancer: Resistance mechanisms and combination therapy strategies.Front Pharmacol. 2022 Aug 25;13:967633. doi: 10.3389/fphar.2022.967633. eCollection 2022. Front Pharmacol. 2022. PMID: 36091750 Free PMC article. Review.
-
The inhibition of PARG attenuates DNA repair in hepatocellular carcinoma.Mol Biomed. 2023 Jan 31;4(1):3. doi: 10.1186/s43556-023-00114-6. Mol Biomed. 2023. PMID: 36719492 Free PMC article. No abstract available.
References
-
- Bürkle A., Poly(ADP-ribose)—The most elaborate metabolite of NAD. FEBS J. 272, 4576–4589 (2005). - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
