

Posttranslational modifications of titin from cardiac muscle: how, where, and what for?
- PMID: 30989819
- PMCID: PMC6850032
- DOI: 10.1111/febs.14854
Posttranslational modifications of titin from cardiac muscle: how, where, and what for?
Abstract
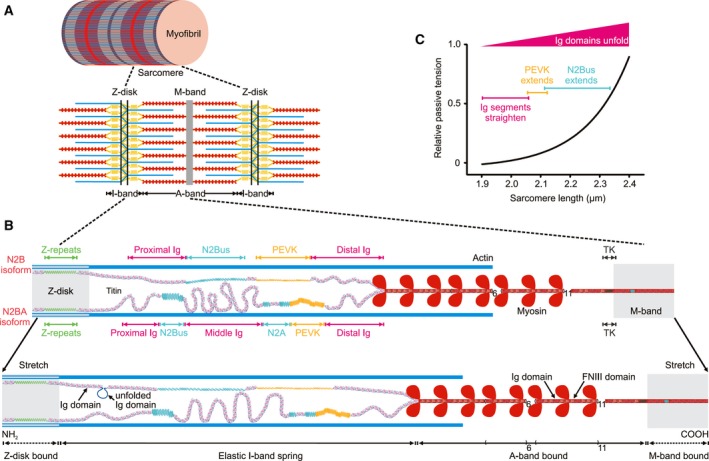
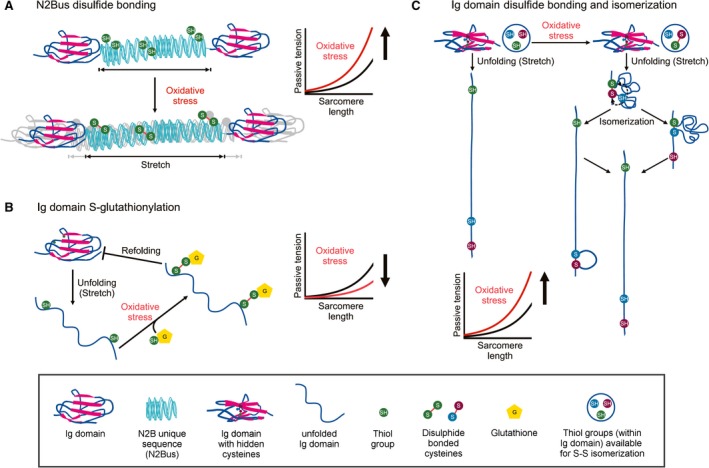
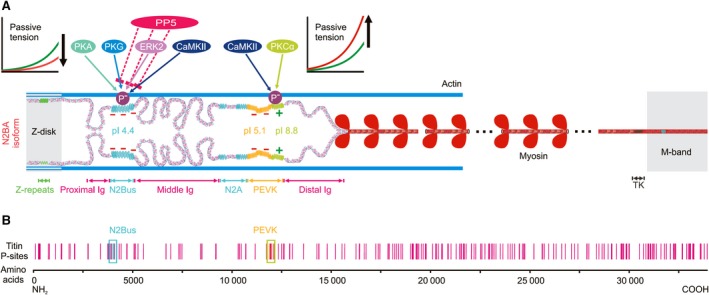
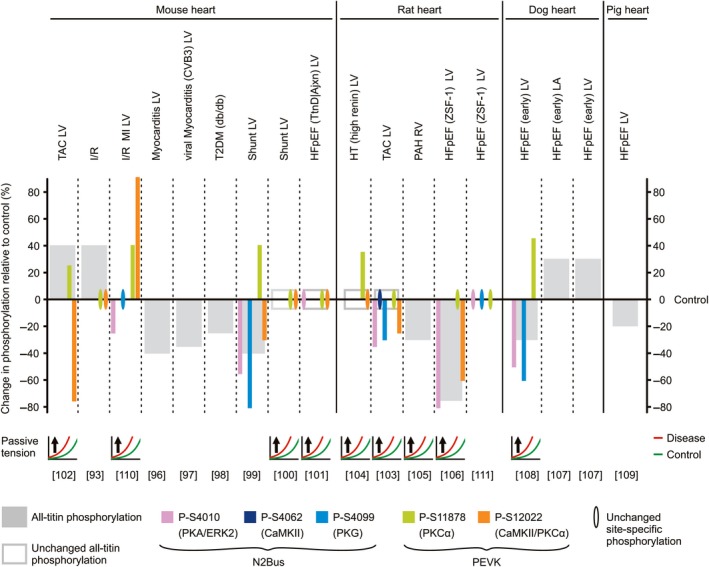
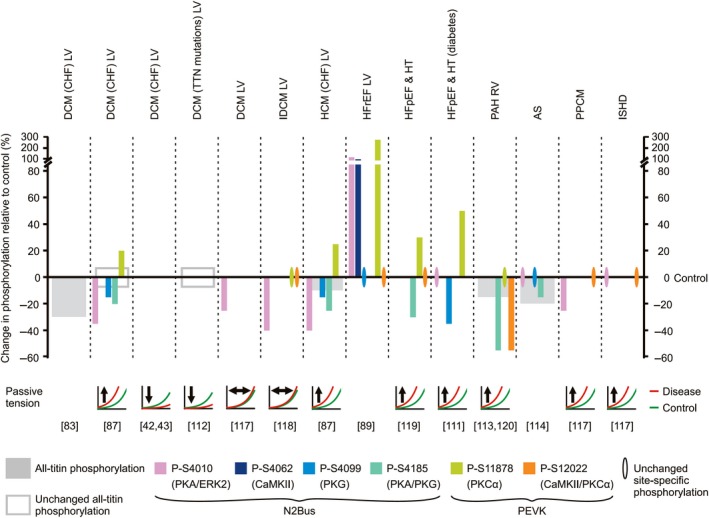
Titin is a giant elastic protein expressed in the contractile units of striated muscle cells, including the sarcomeres of cardiomyocytes. The last decade has seen enormous progress in our understanding of how titin molecular elasticity is modulated in a dynamic manner to help cardiac sarcomeres adjust to the varying hemodynamic demands on the heart. Crucial events mediating the rapid modulation of cardiac titin stiffness are post-translational modifications (PTMs) of titin. In this review, we first recollect what is known from earlier and recent work on the molecular mechanisms of titin extensibility and force generation. The main goal then is to provide a comprehensive overview of current insight into the relationship between titin PTMs and cardiomyocyte stiffness, notably the effect of oxidation and phosphorylation of titin spring segments on titin stiffness. A synopsis is given of which type of oxidative titin modification can cause which effect on titin stiffness. A large part of the review then covers the mechanically relevant phosphorylation sites in titin, their location along the elastic segment, and the protein kinases and phosphatases known to target these sites. We also include a detailed coverage of the complex changes in phosphorylation at specific titin residues, which have been reported in both animal models of heart disease and in human heart failure, and their correlation with titin-based stiffness alterations. Knowledge of the relationship between titin PTMs and titin elasticity can be exploited in the search for therapeutic approaches aimed at softening the pathologically stiffened myocardium in heart failure patients.
Keywords: cytoskeleton; elasticity; heart; heart disease; oxidation; oxidative stress; phosphorylation; protein kinase; protein phosphatase.
© 2019 The Authors. The FEBS Journal published by John Wiley & Sons Ltd on behalf of Federation of European Biochemical Societies.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
References
-
- Linke WA (2008) Sense and stretchability: the role of titin and titin‐associated proteins in myocardial stress‐sensing and mechanical dysfunction. Cardiovasc Res 77, 637–648. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous
