

Tat inhibition by didehydro-Cortistatin A promotes heterochromatin formation at the HIV-1 long terminal repeat
- PMID: 30992052
- PMCID: PMC6466689
- DOI: 10.1186/s13072-019-0267-8
Tat inhibition by didehydro-Cortistatin A promotes heterochromatin formation at the HIV-1 long terminal repeat
Abstract
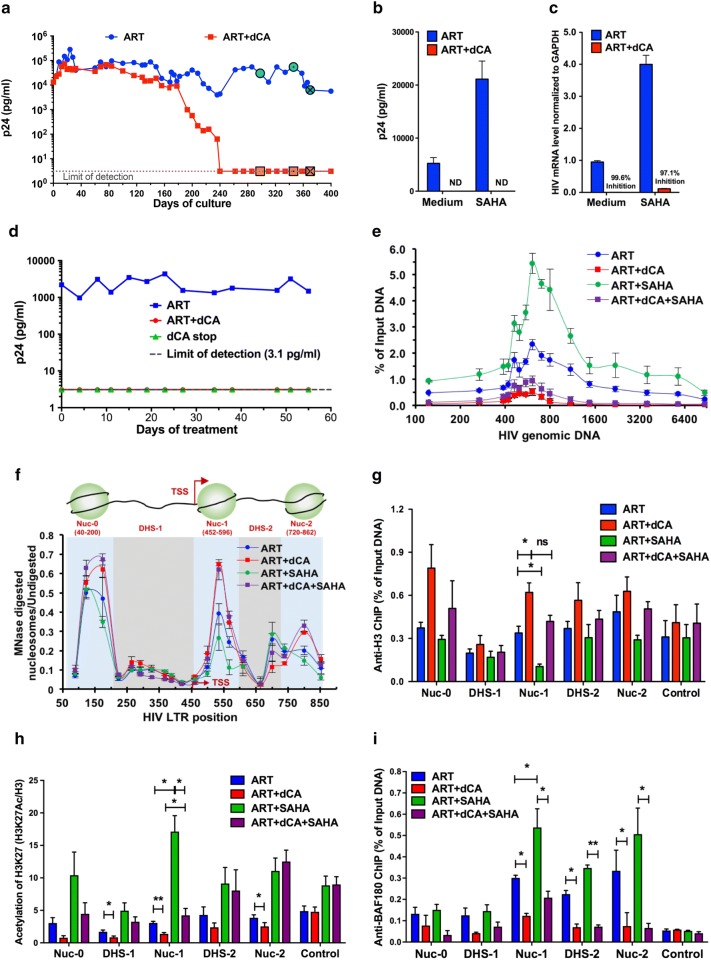
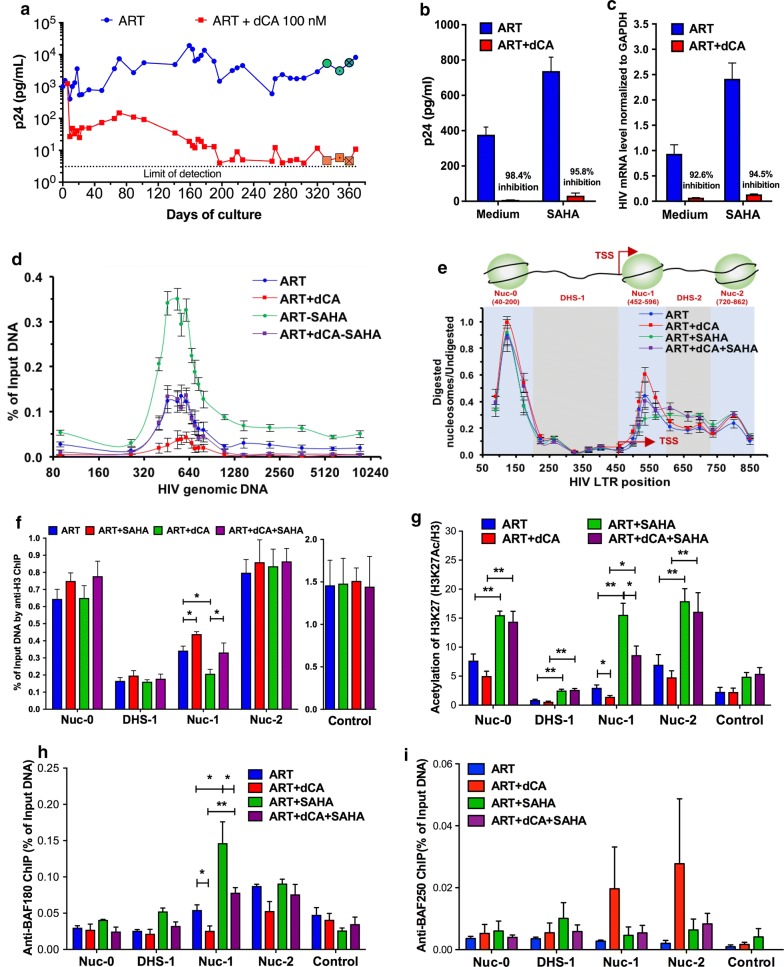
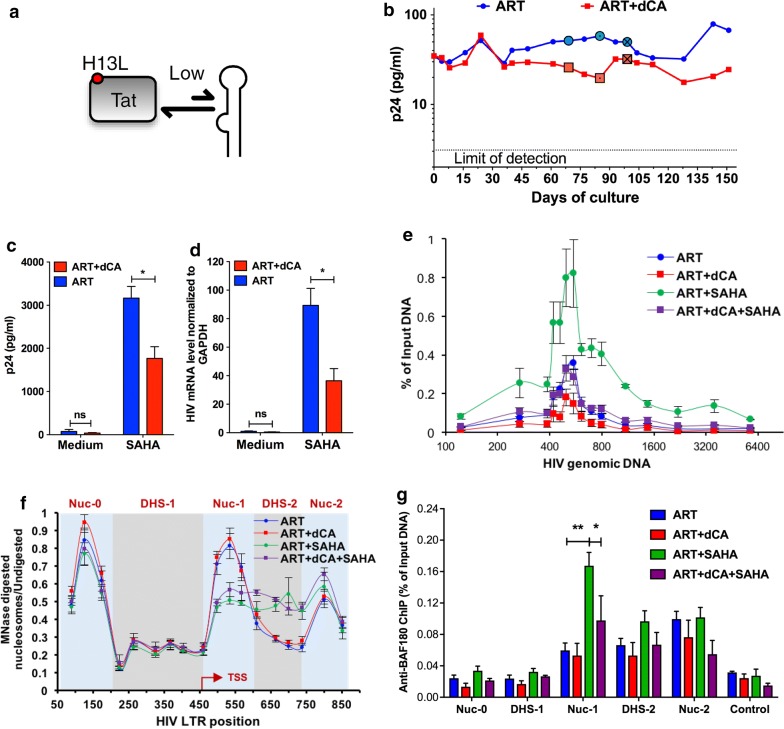
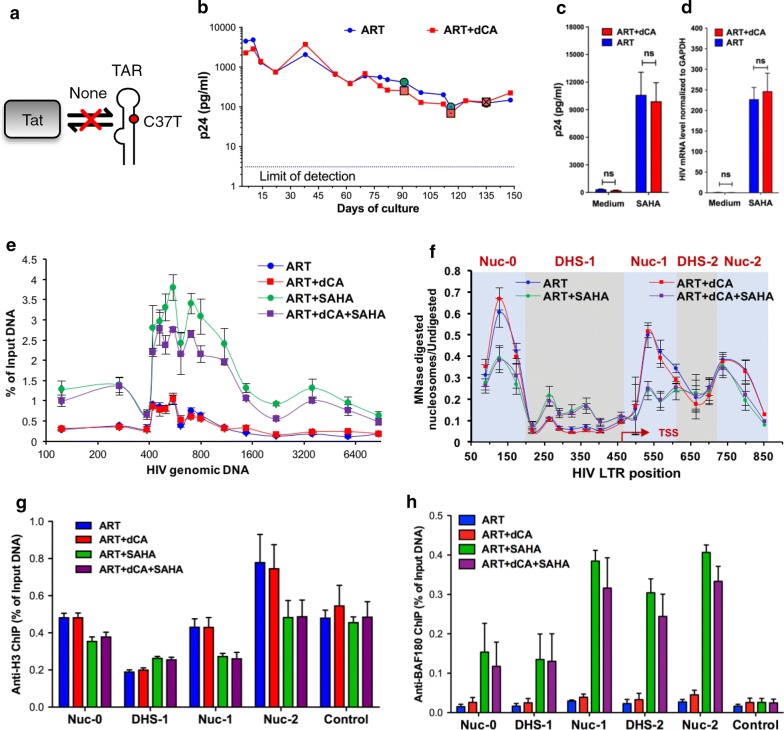
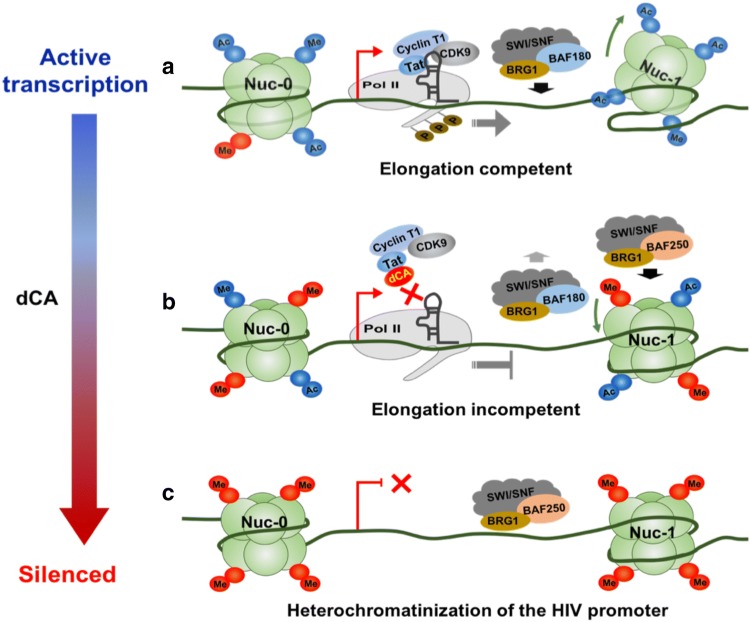
Background: Transcription from the integrated HIV-1 promoter is directly governed by its chromatin environment, and the nucleosome-1 downstream from the transcription start site directly impedes transcription from the HIV-1 promoter. The HIV-1 Tat protein regulates the passage from viral latency to active transcription by binding to the viral mRNA hairpin (TAR) and recruiting transcriptional factors to promote transcriptional elongation. The Tat inhibitor didehydro-Cortistatin A (dCA) inhibits transcription and overtime, the lack of low-grade transcriptional events, triggers epigenetic changes at the latent loci that "lock" HIV transcription in a latent state.
Results: Here we investigated those epigenetic changes using multiple cell line models of HIV-1 latency and active transcription. We demonstrated that dCA treatment does not alter the classic nucleosome positioning at the HIV-1 promoter, but promotes tighter nucleosome/DNA association correlating with increased deacetylated H3 occupancy at nucleosome-1. Recruitment of the SWI/SNF chromatin remodeling complex PBAF, necessary for Tat-mediated transactivation, is also inhibited, while recruitment of the repressive BAF complex is enhanced. These results were supported by loss of RNA polymerase II recruitment on the HIV genome, even during strong stimulation with latency-reversing agents. No epigenetic changes were detected in cell line models of latency with Tat-TAR incompetent proviruses confirming the specificity of dCA for Tat.
Conclusions: We characterized the dCA-mediated epigenetic signature on the HIV genome, which translates into potent blocking effects on HIV expression, further strengthening the potential of Tat inhibitors in "block-and-lock" functional cure approaches.
Conflict of interest statement
The authors declare they have no competing interests.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
