

Annotated bacterial chromosomes from frame-shift-corrected long-read metagenomic data
- PMID: 30992083
- PMCID: PMC6469205
- DOI: 10.1186/s40168-019-0665-y
Annotated bacterial chromosomes from frame-shift-corrected long-read metagenomic data
Abstract
Background: Short-read sequencing technologies have long been the work-horse of microbiome analysis. Continuing technological advances are making the application of long-read sequencing to metagenomic samples increasingly feasible.
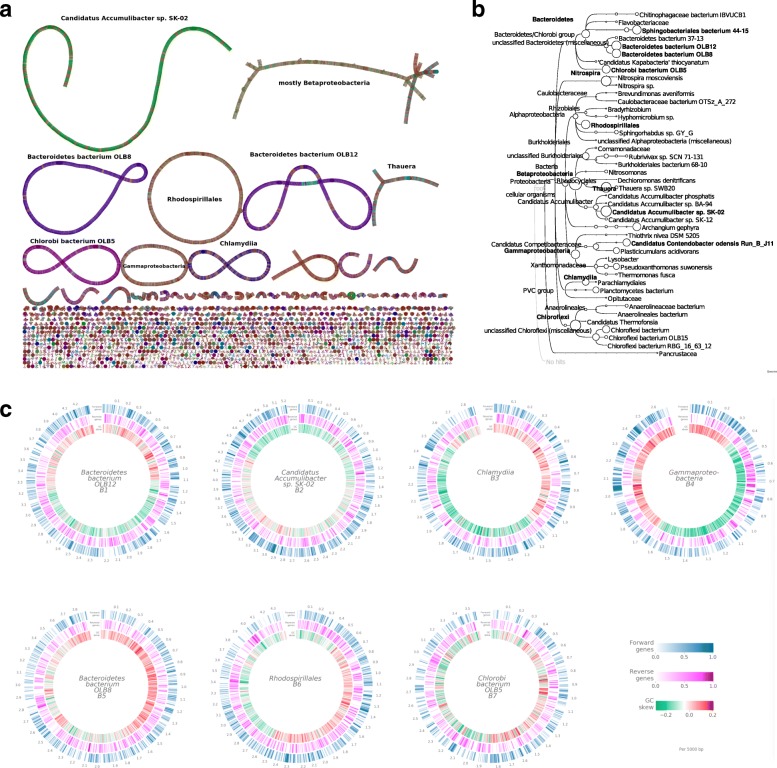
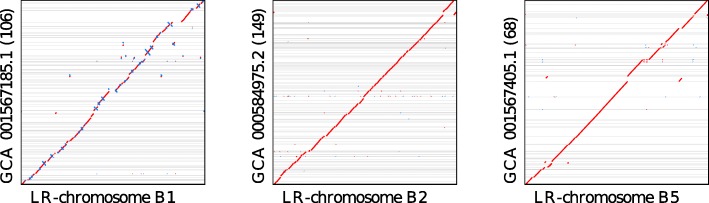
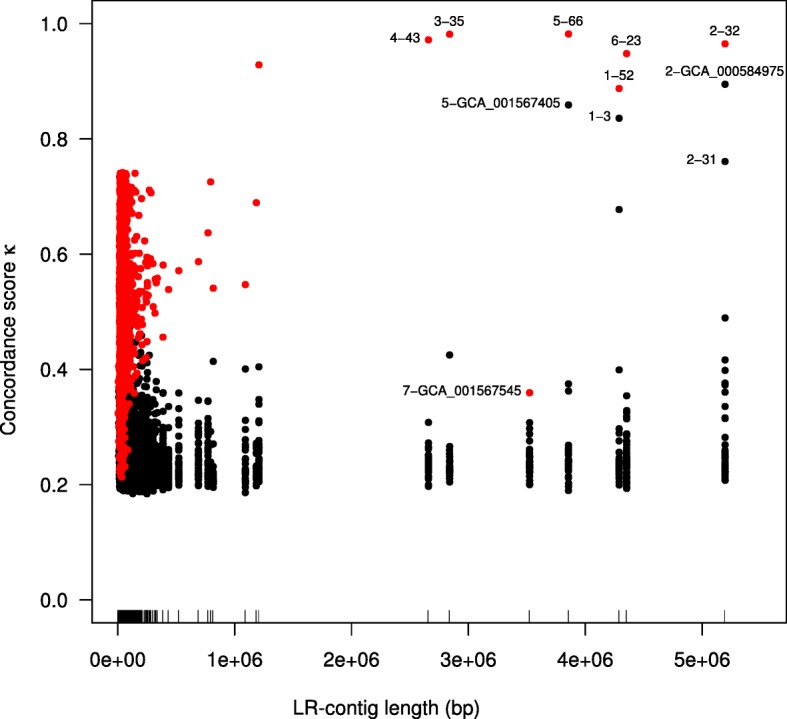
Results: We demonstrate that whole bacterial chromosomes can be obtained from an enriched community, by application of MinION sequencing to a sample from an EBPR bioreactor, producing 6 Gb of sequence that assembles into multiple closed bacterial chromosomes. We provide a simple pipeline for processing such data, which includes a new approach to correcting erroneous frame-shifts.
Conclusions: Advances in long-read sequencing technology and corresponding algorithms will allow the routine extraction of whole chromosomes from environmental samples, providing a more detailed picture of individual members of a microbiome.
Keywords: Algorithms; Frame-shifts; Long-read sequencing; Microbial genomics; Microbiome; Sequence assembly; Software.
Conflict of interest statement
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Figures



References
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
