

Identification of the Bacterial Biosynthetic Gene Clusters of the Oral Microbiome Illuminates the Unexplored Social Language of Bacteria during Health and Disease
- PMID: 30992349
- PMCID: PMC6469967
- DOI: 10.1128/mBio.00321-19
Identification of the Bacterial Biosynthetic Gene Clusters of the Oral Microbiome Illuminates the Unexplored Social Language of Bacteria during Health and Disease
Abstract
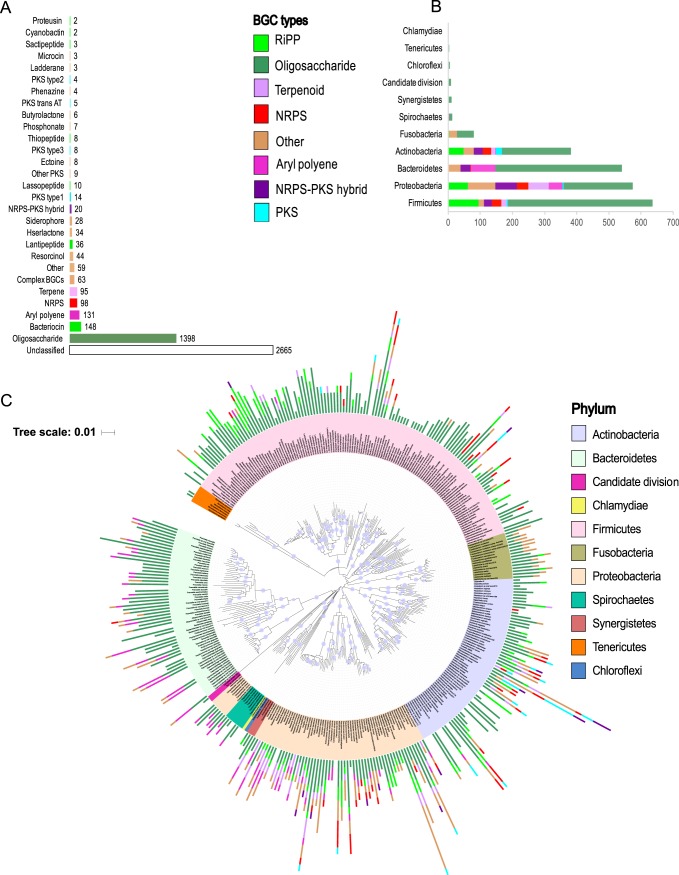
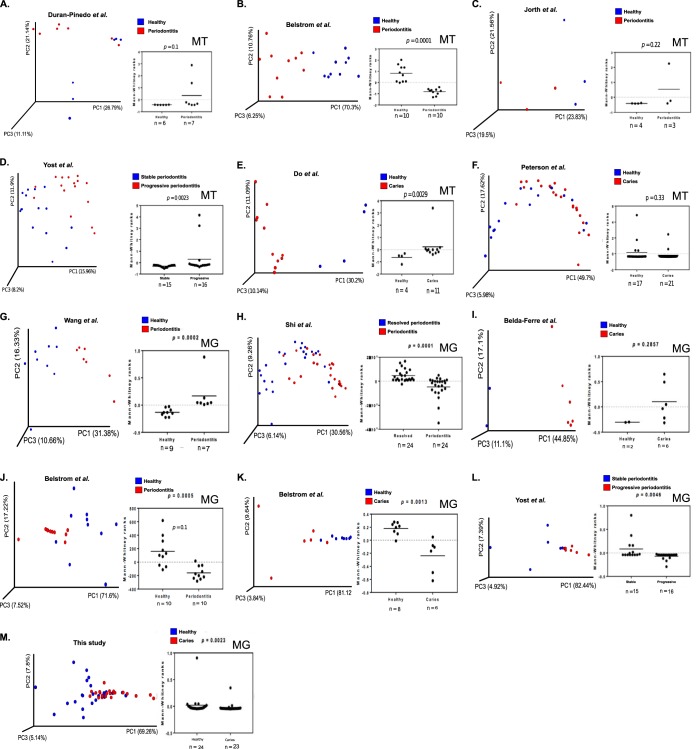
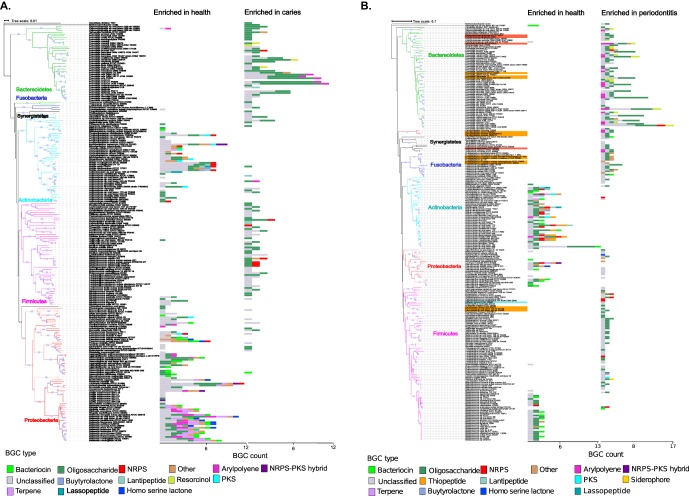
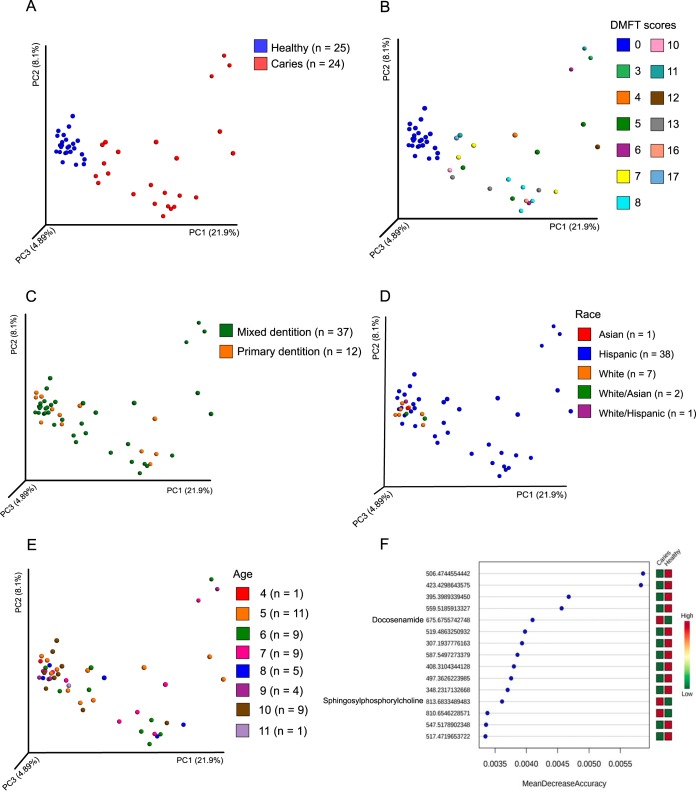
Small molecules are the primary communication media of the microbial world. Recent bioinformatic studies, exploring the biosynthetic gene clusters (BGCs) which produce many small molecules, have highlighted the incredible biochemical potential of the signaling molecules encoded by the human microbiome. Thus far, most research efforts have focused on understanding the social language of the gut microbiome, leaving crucial signaling molecules produced by oral bacteria and their connection to health versus disease in need of investigation. In this study, a total of 4,915 BGCs were identified across 461 genomes representing a broad taxonomic diversity of oral bacteria. Sequence similarity networking provided a putative product class for more than 100 unclassified novel BGCs. The newly identified BGCs were cross-referenced against 254 metagenomes and metatranscriptomes derived from individuals either with good oral health or with dental caries or periodontitis. This analysis revealed 2,473 BGCs, which were differentially represented across the oral microbiomes associated with health versus disease. Coabundance network analysis identified numerous inverse correlations between BGCs and specific oral taxa. These correlations were present in healthy individuals but greatly reduced in individuals with dental caries, which may suggest a defect in colonization resistance. Finally, corroborating mass spectrometry identified several compounds with homology to products of the predicted BGC classes. Together, these findings greatly expand the number of known biosynthetic pathways present in the oral microbiome and provide an atlas for experimental characterization of these abundant, yet poorly understood, molecules and socio-chemical relationships, which impact the development of caries and periodontitis, two of the world's most common chronic diseases.IMPORTANCE The healthy oral microbiome is symbiotic with the human host, importantly providing colonization resistance against potential pathogens. Dental caries and periodontitis are two of the world's most common and costly chronic infectious diseases and are caused by a localized dysbiosis of the oral microbiome. Bacterially produced small molecules, often encoded by BGCs, are the primary communication media of bacterial communities and play a crucial, yet largely unknown, role in the transition from health to dysbiosis. This study provides a comprehensive mapping of the BGC repertoire of the human oral microbiome and identifies major differences in health compared to disease. Furthermore, BGC representation and expression is linked to the abundance of particular oral bacterial taxa in health versus dental caries and periodontitis. Overall, this study provides a significant insight into the chemical communication network of the healthy oral microbiome and how it devolves in the case of two prominent diseases.
Keywords: biosynthetic gene clusters; caries; genome mining; oral microbiome; periodontitis; small molecules.
Copyright © 2019 Aleti et al.
Figures


References
-
- Donia MS, Cimermancic P, Schulze CJ, Wieland Brown LC, Martin J, Mitreva M, Clardy J, Linington RG, Fischbach MA. 2014. A systematic analysis of biosynthetic gene clusters in the human microbiome reveals a common family of antibiotics. Cell 158:1402–1414. doi: 10.1016/j.cell.2014.08.032. - DOI - PMC - PubMed
-
- Cimermancic P, Medema MH, Claesen J, Kurita K, Wieland Brown LC, Mavrommatis K, Pati A, Godfrey PA, Koehrsen M, Clardy J, Birren BW, Takano E, Sali A, Linington RG, Fischbach MA. 2014. Insights into secondary metabolism from a global analysis of prokaryotic biosynthetic gene clusters. Cell 158:412–421. doi: 10.1016/j.cell.2014.06.034. - DOI - PMC - PubMed
-
- Zipperer A, Konnerth MC, Laux C, Berscheid A, Janek D, Weidenmaier C, Burian M, Schilling NA, Slavetinsky C, Marschal M, Willmann M, Kalbacher H, Schittek B, Brotz-Oesterhelt H, Grond S, Peschel A, Krismer B. 2016. Human commensals producing a novel antibiotic impair pathogen colonization. Nature 535:511–516. doi: 10.1038/nature18634. - DOI - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
