

Evolutionary and Genomic Insights into Clostridioides difficile Sequence Type 11: a Diverse Zoonotic and Antimicrobial-Resistant Lineage of Global One Health Importance
- PMID: 30992351
- PMCID: PMC6469969
- DOI: 10.1128/mBio.00446-19
Evolutionary and Genomic Insights into Clostridioides difficile Sequence Type 11: a Diverse Zoonotic and Antimicrobial-Resistant Lineage of Global One Health Importance
Abstract
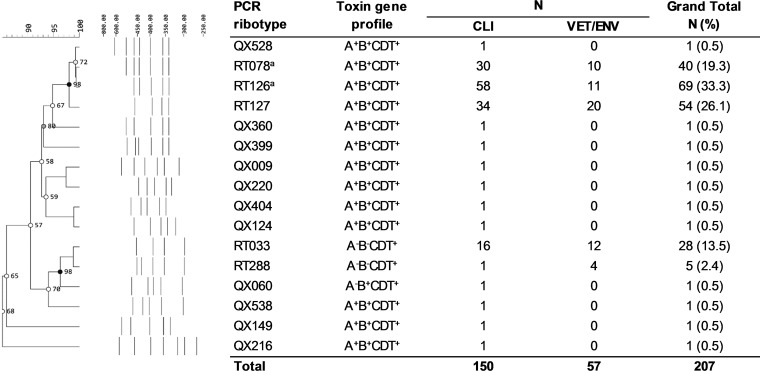
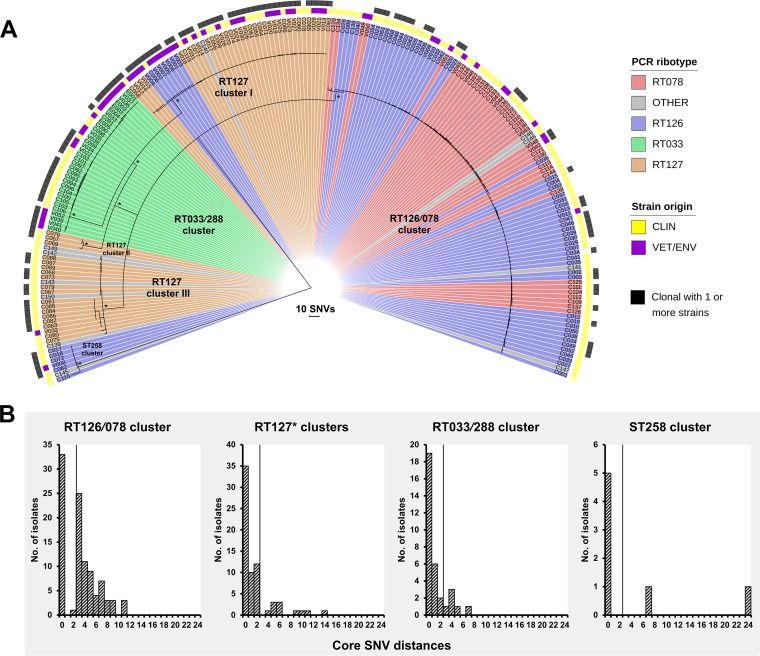
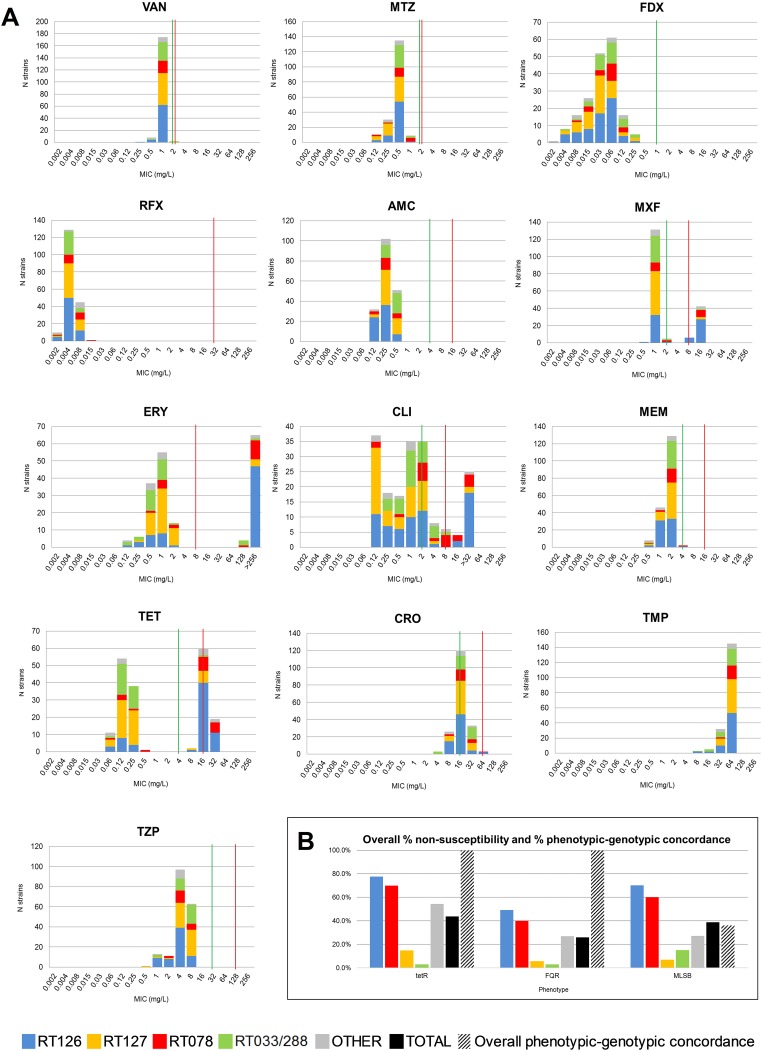
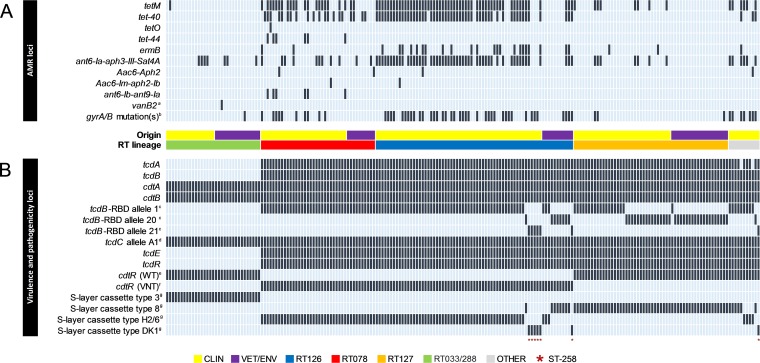
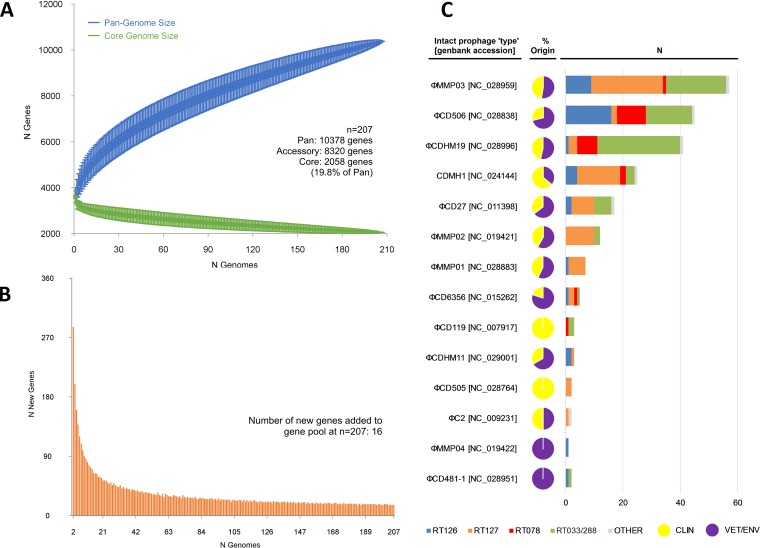
Clostridioides difficile (Clostridium difficile) sequence type 11 (ST11) is well established in production animal populations worldwide and contributes considerably to the global burden of C. difficile infection (CDI) in humans. Increasing evidence of shared ancestry and genetic overlap of PCR ribotype 078 (RT078), the most common ST11 sublineage, between human and animal populations suggests that CDI may be a zoonosis. We performed whole-genome sequencing (WGS) on a collection of 207 ST11 and closely related ST258 isolates of human and veterinary/environmental origin, comprising 16 RTs collected from Australia, Asia, Europe, and North America. Core genome single nucleotide variant (SNV) analysis identified multiple intraspecies and interspecies clonal groups (isolates separated by ≤2 core genome SNVs) in all the major RT sublineages: 078, 126, 127, 033, and 288. Clonal groups comprised isolates spread across different states, countries, and continents, indicative of reciprocal long-range dissemination and possible zoonotic/anthroponotic transmission. Antimicrobial resistance genotypes and phenotypes varied across host species, geographic regions, and RTs and included macrolide/lincosamide resistance (Tn6194 [ermB]), tetracycline resistance (Tn6190 [tetM] and Tn6164 [tet44]), and fluoroquinolone resistance (gyrA/B mutations), as well as numerous aminoglycoside resistance cassettes. The population was defined by a large "open" pan-genome (10,378 genes), a remarkably small core genome of 2,058 genes (only 19.8% of the gene pool), and an accessory genome containing a large and diverse collection of important prophages of the Siphoviridae and Myoviridae This study provides novel insights into strain relatedness and genetic variability of C. difficile ST11, a lineage of global One Health importance.IMPORTANCE Historically, Clostridioides difficile (Clostridium difficile) has been associated with life-threatening diarrhea in hospitalized patients. Increasing rates of C. difficile infection (CDI) in the community suggest exposure to C. difficile reservoirs outside the hospital, including animals, the environment, or food. C. difficile sequence type 11 (ST11) is known to infect/colonize livestock worldwide and comprises multiple ribotypes, many of which cause disease in humans, suggesting CDI may be a zoonosis. Using high-resolution genomics, we investigated the evolution and zoonotic potential of ST11 and a new closely related ST258 lineage sourced from diverse origins. We found multiple intra- and interspecies clonal transmission events in all ribotype sublineages. Clones were spread across multiple continents, often without any health care association, indicative of zoonotic/anthroponotic long-range dissemination in the community. ST11 possesses a massive pan-genome and numerous clinically important antimicrobial resistance elements and prophages, which likely contribute to the success of this globally disseminated lineage of One Health importance.
Keywords: Clostridium difficile; One Health; antimicrobial resistance; epidemiology; evolution; livestock; microbial genomics; toxin; transmission; zoonosis.
© Crown copyright 2019.
Figures
References
-
- Knetsch CW, Connor TR, Mutreja A, van Dorp SM, Sanders IM, Browne HP, Harris D, Lipman L, Keessen EC, Corver J, Kuijper EJ, Lawley TD. 2014. Whole genome sequencing reveals potential spread of Clostridium difficile between humans and farm animals in the Netherlands, 2002 to 2011. Euro Surveill 19:30–41. - PMC - PubMed
-
- Knight DR, Squire MM, Collins DA, Riley TV. 2016. Genome analysis of Clostridium difficile PCR ribotype 014 lineage in Australian pigs and humans reveals a diverse genetic repertoire and signatures of long-range interspecies transmission. Front Microbiol 7:2138. doi: 10.3389/fmicb.2016.02138. - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
Associated data
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
