

The role of platelets in mediating a response to human influenza infection
- PMID: 30992428
- PMCID: PMC6467905
- DOI: 10.1038/s41467-019-09607-x
The role of platelets in mediating a response to human influenza infection
Abstract
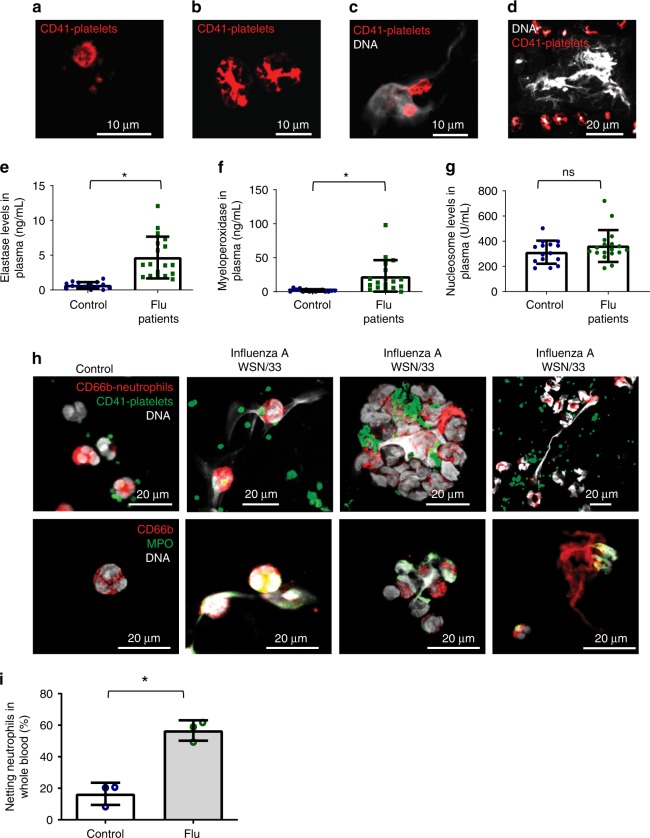
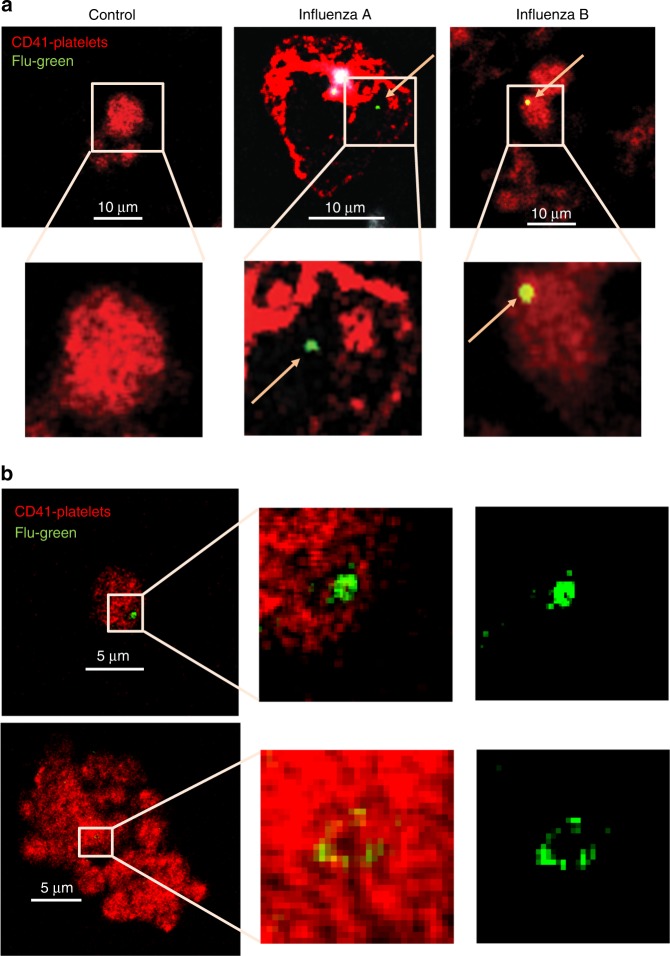
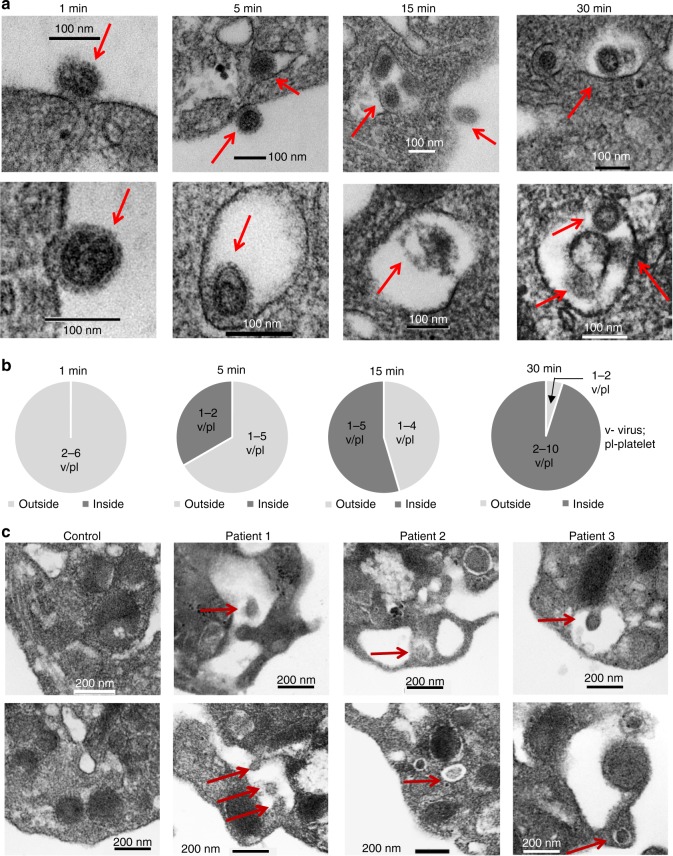
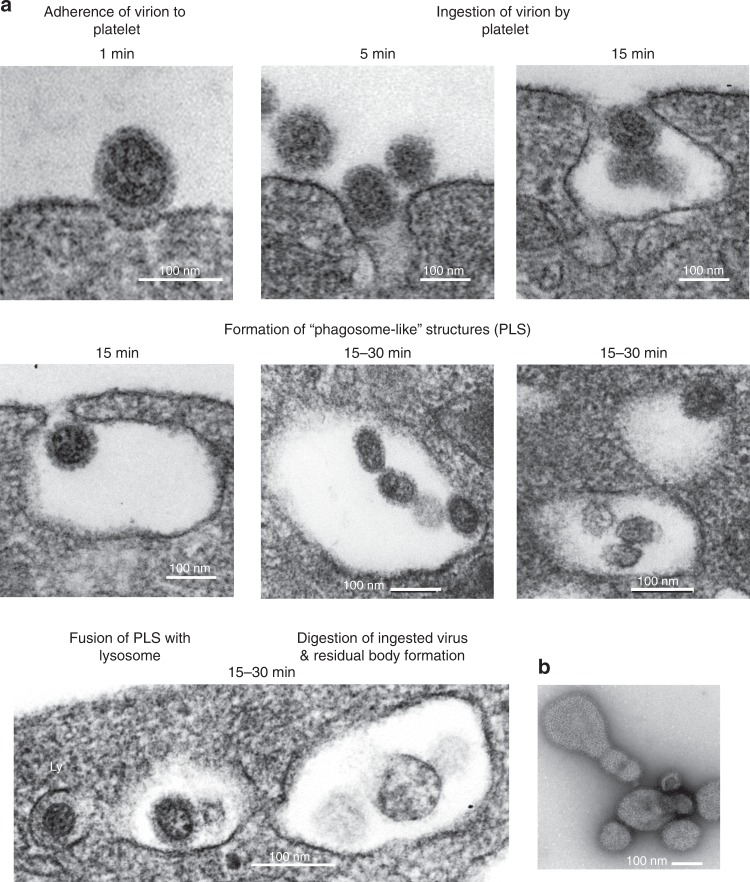
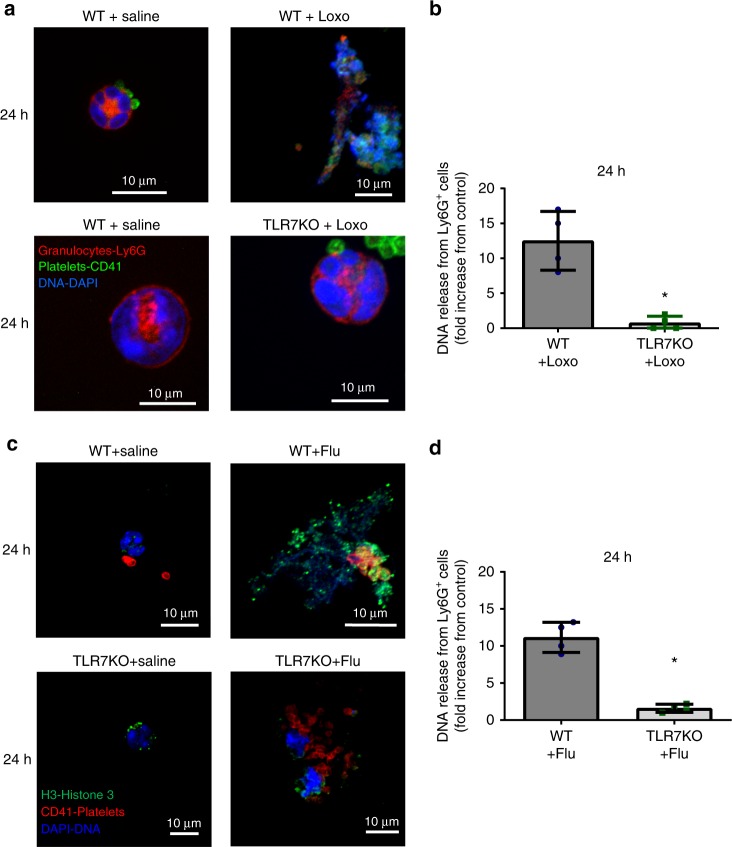
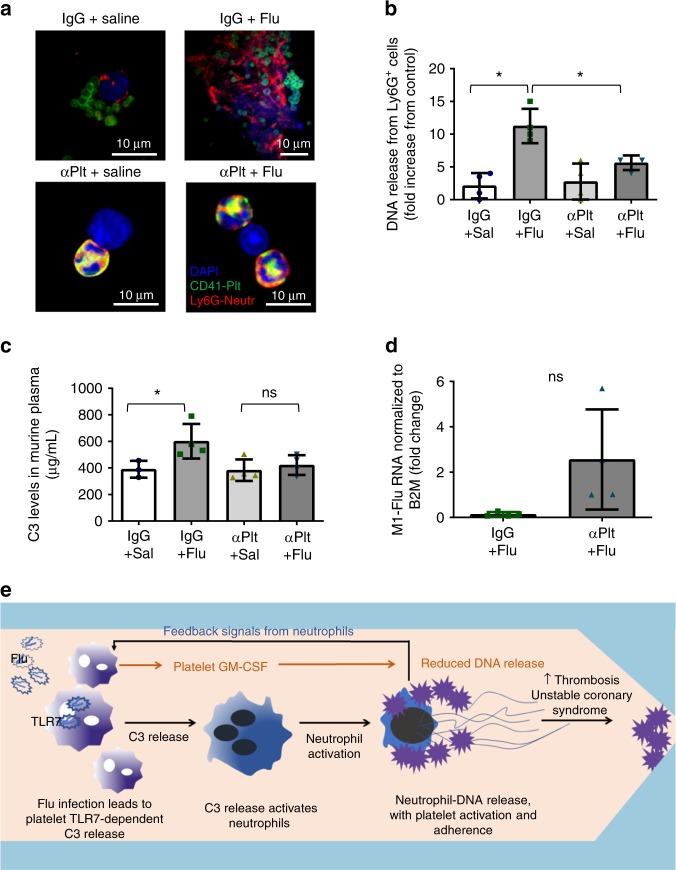
Influenza infection increases the incidence of myocardial infarction but the reason is unknown. Platelets mediate vascular occlusion through thrombotic functions but are also recognized to have immunomodulatory activity. To determine if platelet processes are activated during influenza infection, we collected blood from 18 patients with acute influenza infection. Microscopy reveals activated platelets, many containing viral particles and extracellular-DNA associated with platelets. To understand the mechanism, we isolate human platelets and treat them with influenza A virus. Viral-engulfment leads to C3 release from platelets as a function of TLR7 and C3 leads to neutrophil-DNA release and aggregation. TLR7 specificity is confirmed in murine models lacking the receptor, and platelet depletion models support platelet-mediated C3 and neutrophil-DNA release post-influenza infection. These findings demonstrate that the initial intrinsic defense against influenza is mediated by platelet-neutrophil cross-communication that tightly regulates host immune and complement responses but can also lead to thrombotic vascular occlusion.
Conflict of interest statement
The authors declare no competing interests.
Figures
References
-
- Center for Disease Control. CDC WONDER. Underlying Cause of Death 1999–2016. https://wonder.cdc.gov/ucd-icd10.html (1999–2016).
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Miscellaneous
