

Polygenic prediction via Bayesian regression and continuous shrinkage priors
- PMID: 30992449
- PMCID: PMC6467998
- DOI: 10.1038/s41467-019-09718-5
Polygenic prediction via Bayesian regression and continuous shrinkage priors
Abstract
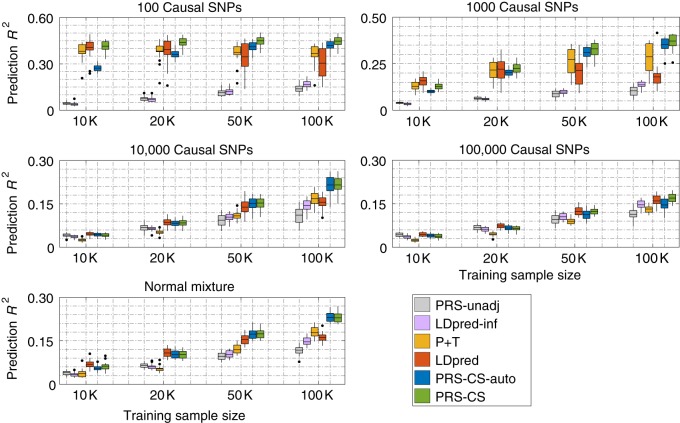
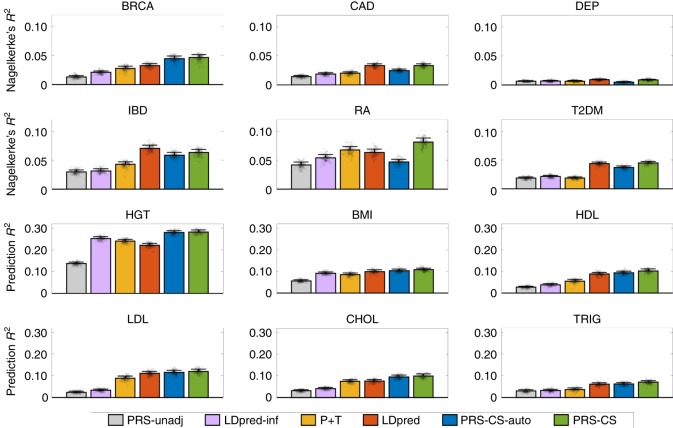
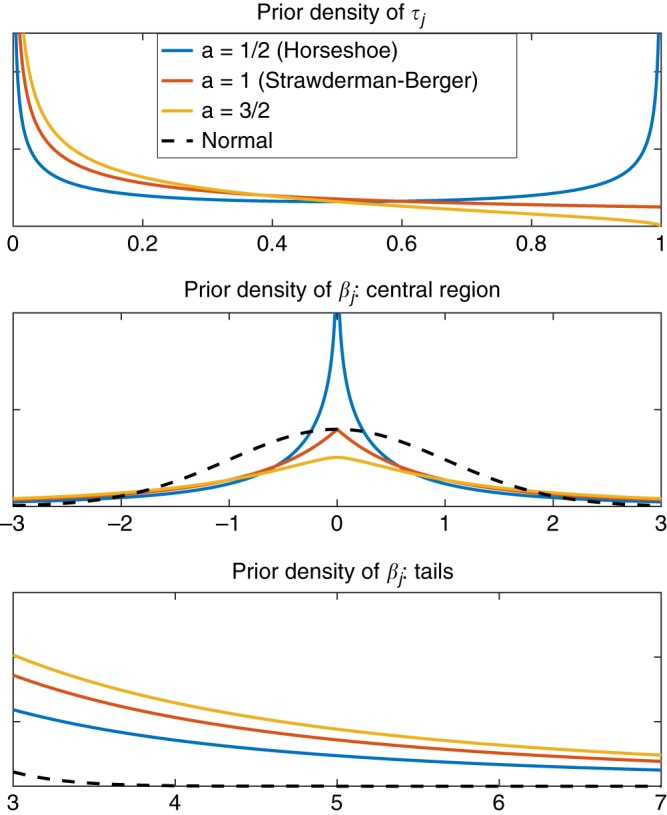
Polygenic risk scores (PRS) have shown promise in predicting human complex traits and diseases. Here, we present PRS-CS, a polygenic prediction method that infers posterior effect sizes of single nucleotide polymorphisms (SNPs) using genome-wide association summary statistics and an external linkage disequilibrium (LD) reference panel. PRS-CS utilizes a high-dimensional Bayesian regression framework, and is distinct from previous work by placing a continuous shrinkage (CS) prior on SNP effect sizes, which is robust to varying genetic architectures, provides substantial computational advantages, and enables multivariate modeling of local LD patterns. Simulation studies using data from the UK Biobank show that PRS-CS outperforms existing methods across a wide range of genetic architectures, especially when the training sample size is large. We apply PRS-CS to predict six common complex diseases and six quantitative traits in the Partners HealthCare Biobank, and further demonstrate the improvement of PRS-CS in prediction accuracy over alternative methods.
Conflict of interest statement
The authors declare no competing interests.
Figures
References
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
