

Gene discovery informatics toolkit defines candidate genes for unexplained infertility and prenatal or infantile mortality
- PMID: 30993004
- PMCID: PMC6465277
- DOI: 10.1038/s41525-019-0081-z
Gene discovery informatics toolkit defines candidate genes for unexplained infertility and prenatal or infantile mortality
Abstract
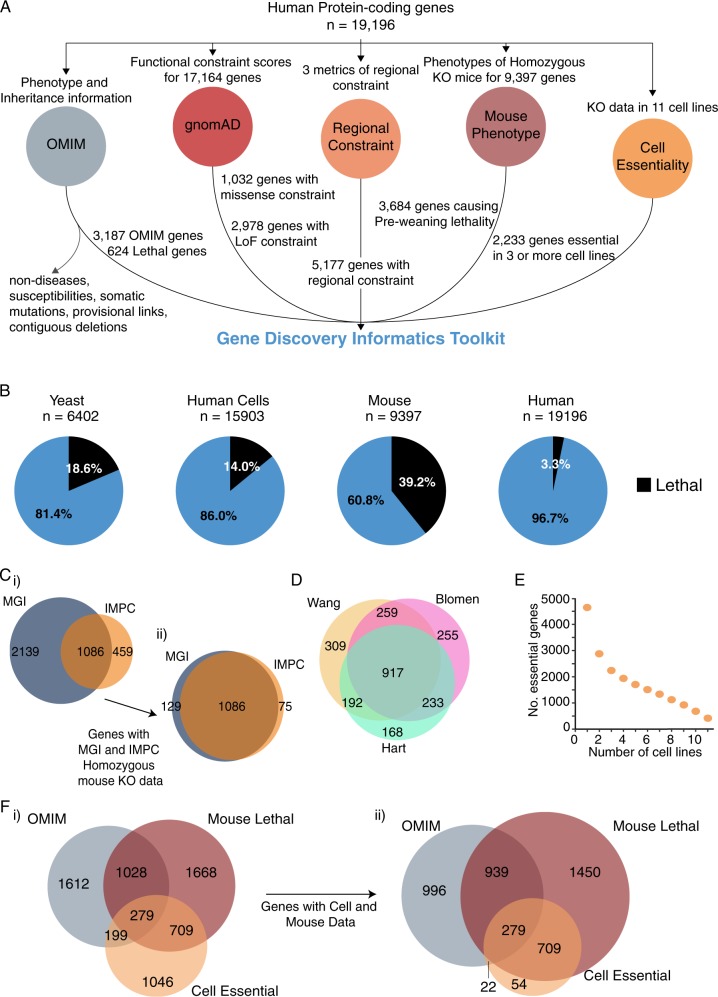
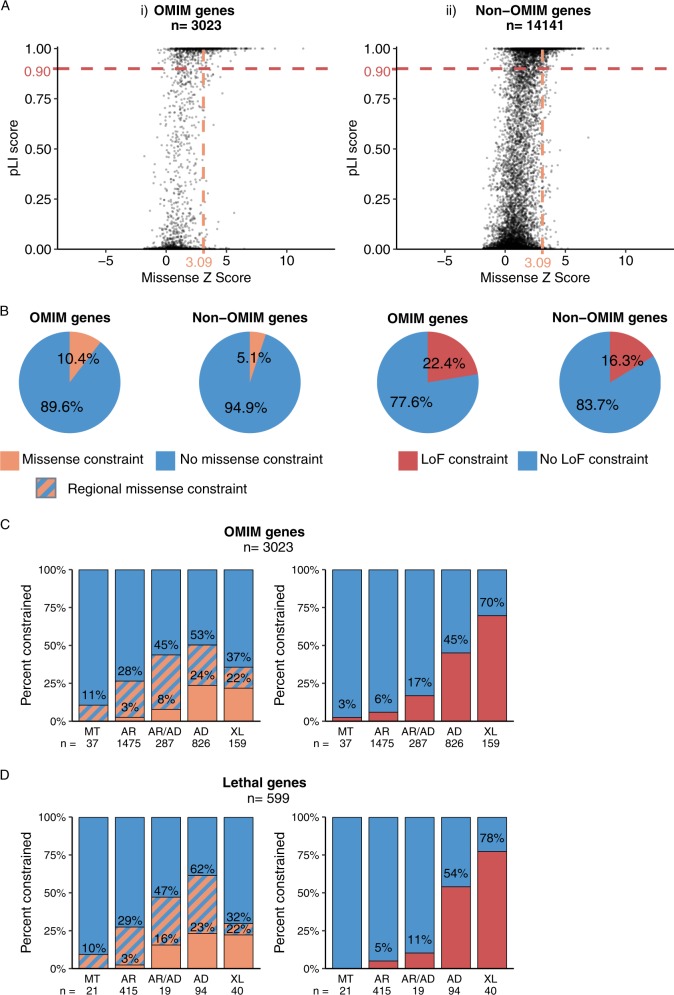
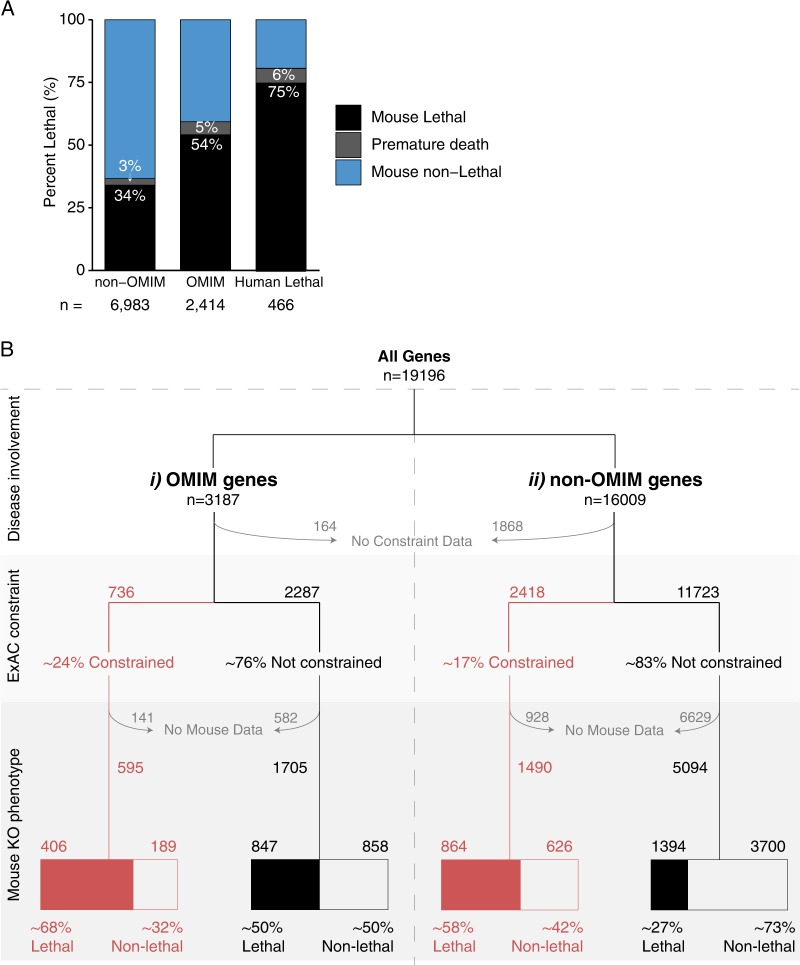
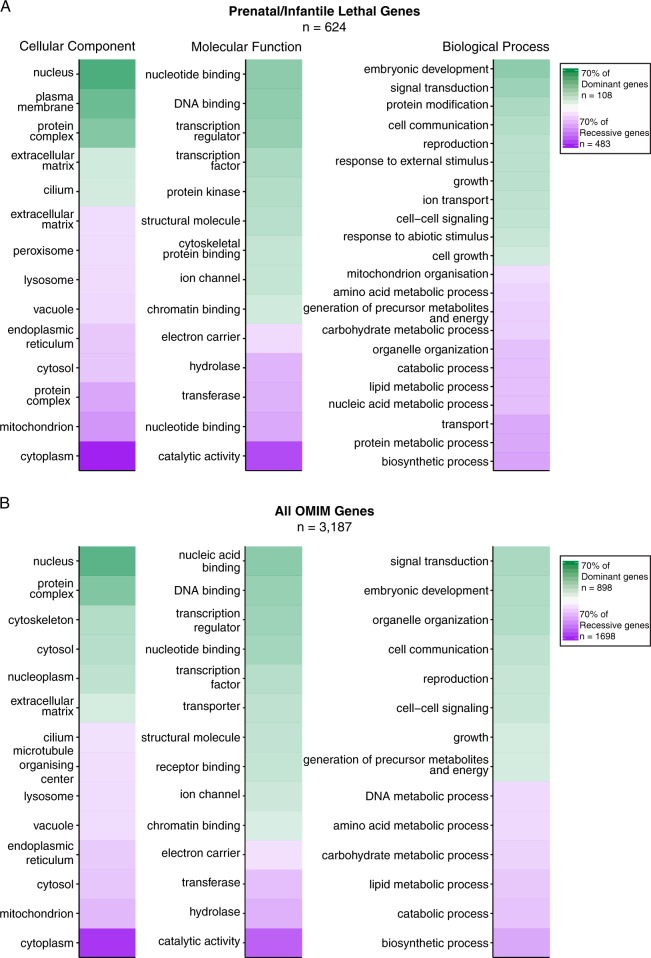
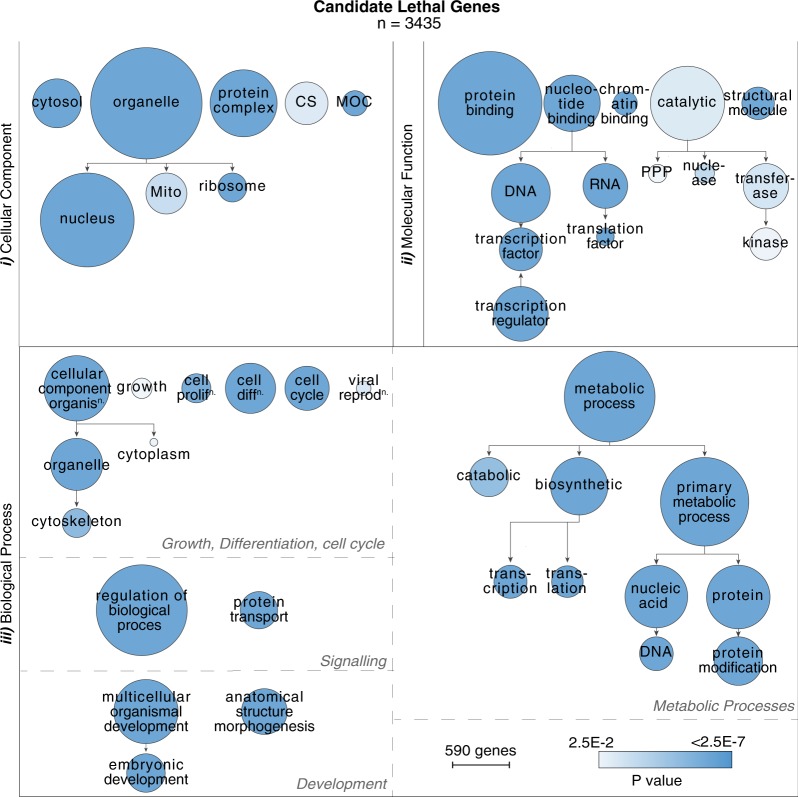
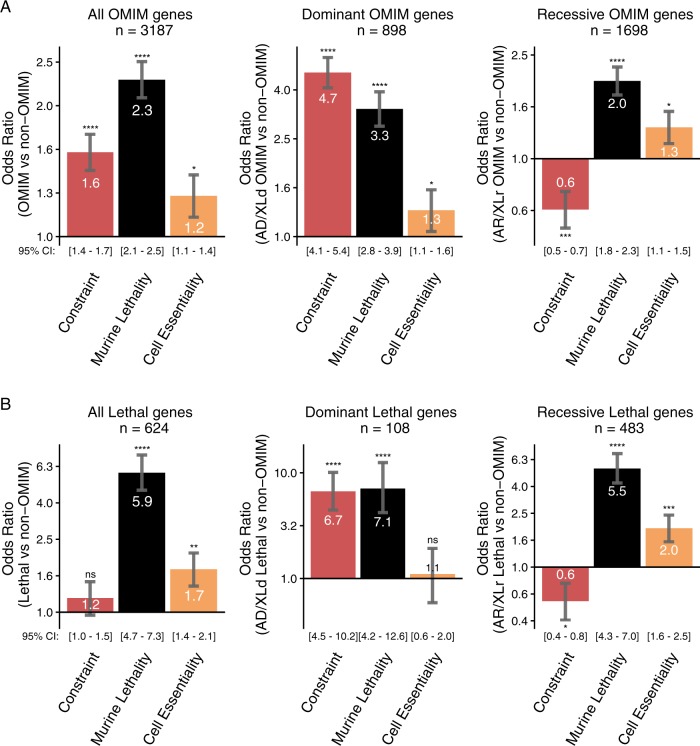
Despite a recent surge in novel gene discovery, genetic causes of prenatal-lethal phenotypes remain poorly defined. To advance gene discovery in prenatal-lethal disorders, we created an easy-to-mine database integrating known human phenotypes with inheritance pattern, scores of genetic constraint, and murine and cellular knockout phenotypes-then critically assessed defining features of known prenatal-lethal genes, among 3187 OMIM genes, and relative to 16,009 non-disease genes. While around one-third (39%) of protein-coding genes are essential for murine development, we curate only 3% (624) of human protein-coding genes linked currently to prenatal/infantile lethal disorders. 75% prenatal-lethal genes are linked to developmental lethality in knockout mice, compared to 54% for all OMIM genes and 34% among non-disease genes. Genetic constraint correlates with inheritance pattern (autosomal recessive <<autosomal dominant <X-linked), and is greatest among prenatal-lethal genes. Importantly, >90% of recessive genes show neither missense nor loss-of-function constraint, even for prenatal-lethal genes. Detailed ontology mapping for 624 prenatal-lethal genes shows marked enrichment among dominant genes for nuclear proteins with roles in RNA/DNA biology, with recessive genes enriched in cytoplasmic (mitochondrial) metabolic proteins. We conclude that genes without genetic constraint should not be excluded as potential novel disease genes, and especially for recessive conditions (<10% constrained). Prenatal lethal genes are 5.9-fold more likely to be associated with a lethal murine phenotype than non-disease genes. Cell essential genes are largely a subset of mouse-lethal genes, notably under-represented among known OMIM genes, and strong candidates for gamete/embryo non-viability. We therefore curate 3435 'candidate developmental lethal' human genes: essential for murine development or cellular viability, not yet linked to human disorders, presenting strong candidates for unexplained infertility and prenatal/infantile mortality.
Conflict of interest statement
S.T.C. is director of Frontier Genomics Pty Ltd (Australia). Frontier Genomics has not traded (as of November 12th, 2018). Frontier Genomics Pty Ltd (Australia) will not benefit from publication of these data. The remaining authors declare no competing interests.
Figures
References
-
- Samocha, K. E. et al. Regional missense constraint improves variant deleteriousness prediction. https://www.biorxiv.org/content/10.1101/148353v1 (2017). - DOI
Publication types
LinkOut - more resources
Full Text Sources
