

Pulsatile MEK Inhibition Improves Anti-tumor Immunity and T Cell Function in Murine Kras Mutant Lung Cancer
- PMID: 30995478
- PMCID: PMC6719696
- DOI: 10.1016/j.celrep.2019.03.066
Pulsatile MEK Inhibition Improves Anti-tumor Immunity and T Cell Function in Murine Kras Mutant Lung Cancer
Abstract
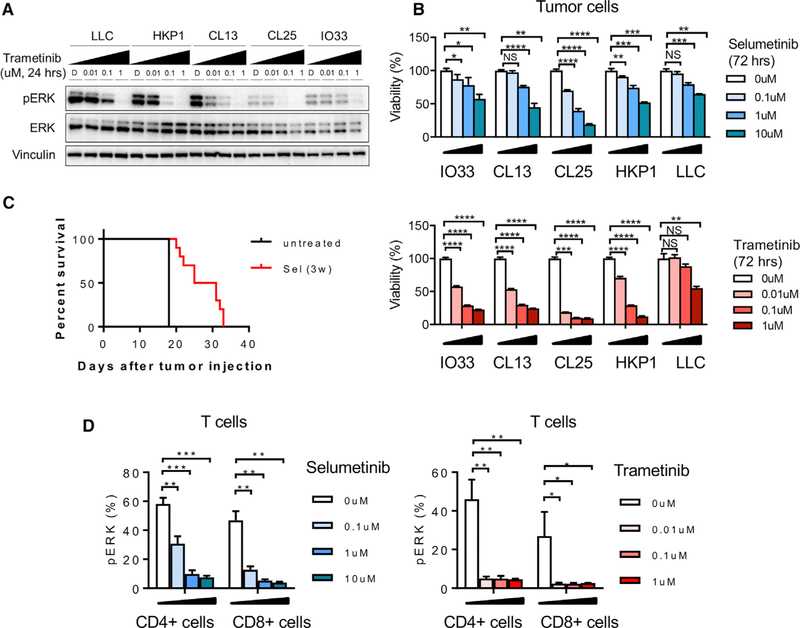
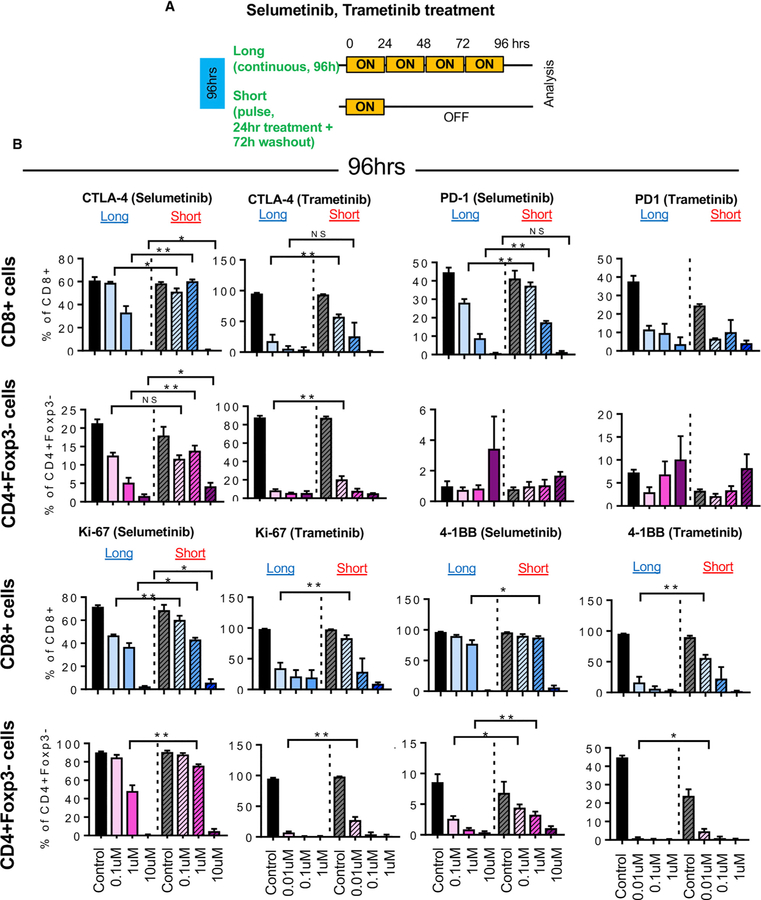
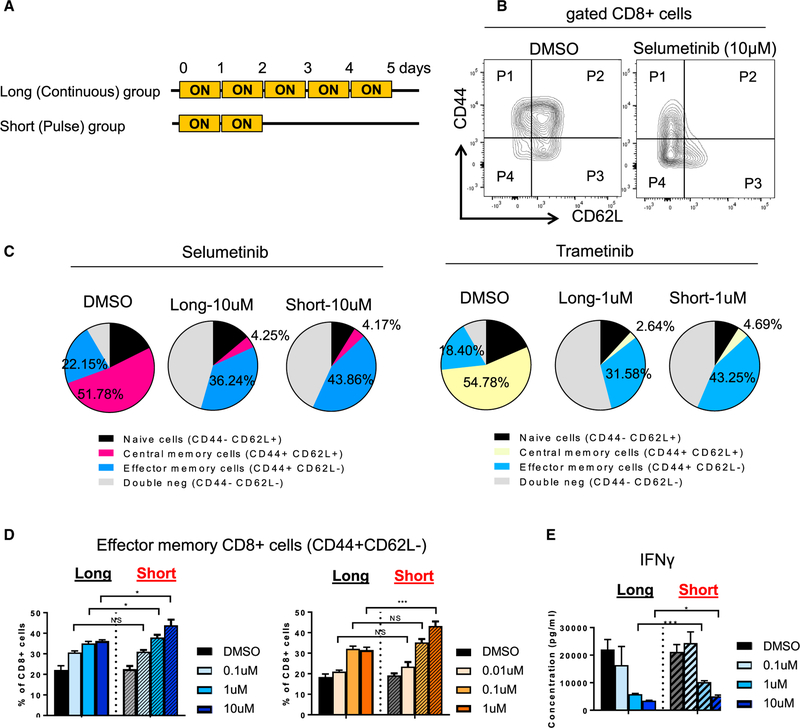
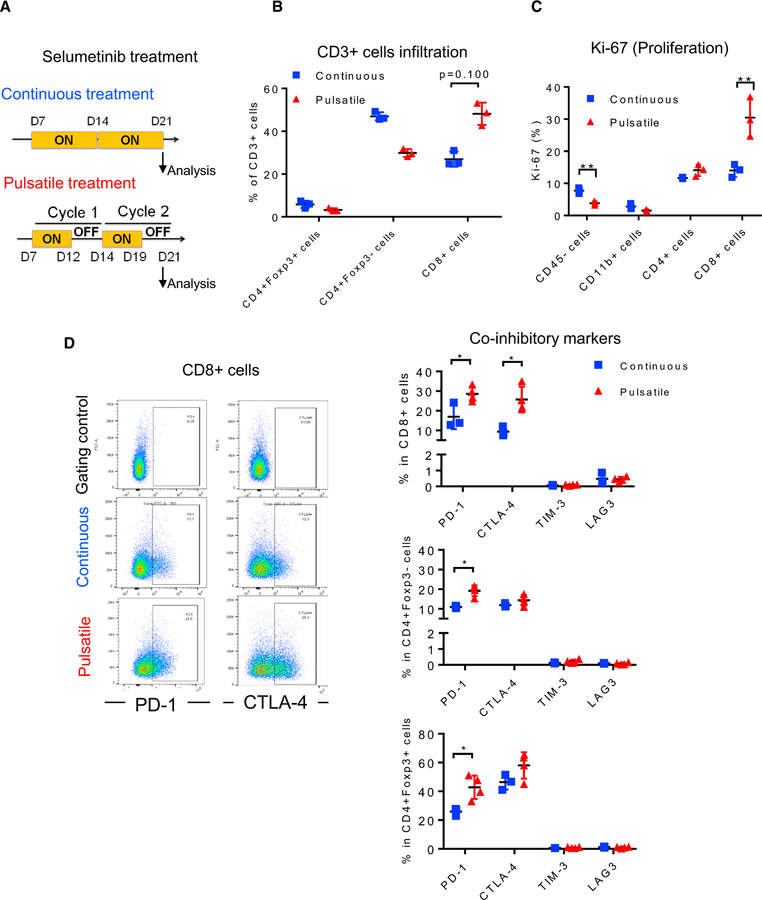
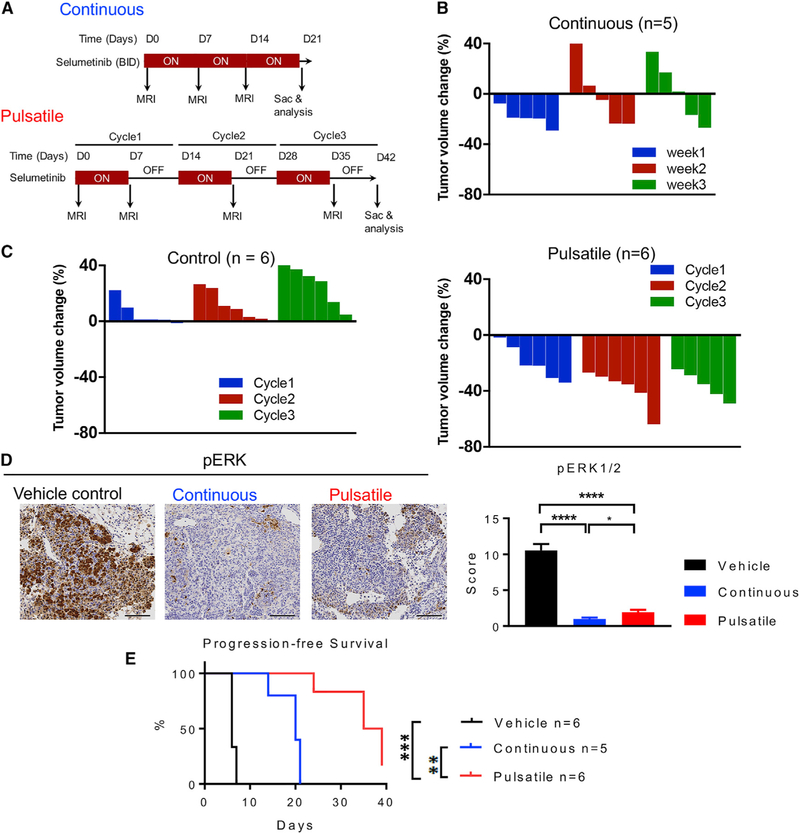
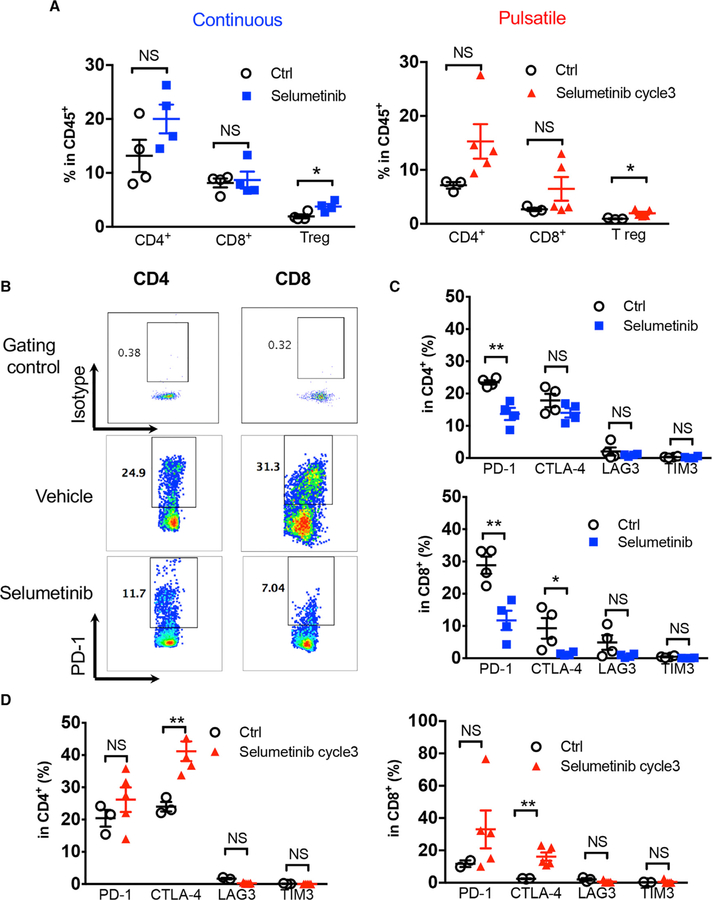
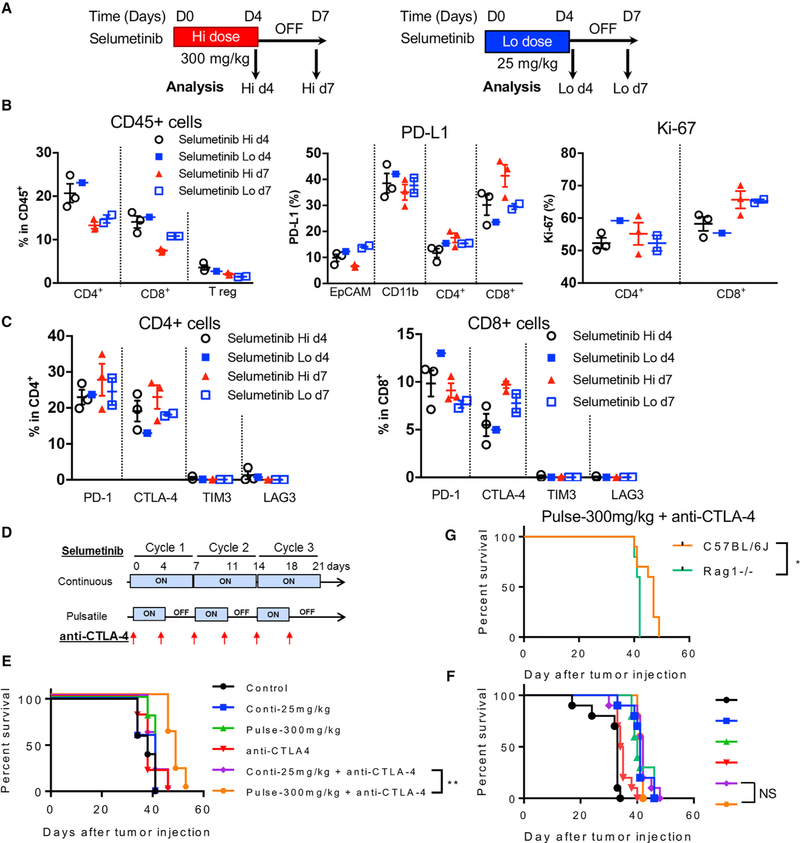
KRAS is one of the driver oncogenes in non-small-cell lung cancer (NSCLC) but remains refractory to current modalities of targeted pathway inhibition, which include inhibiting downstream kinase MEK to circumvent KRAS activation. Here, we show that pulsatile, rather than continuous, treatment with MEK inhibitors (MEKis) maintains T cell activation and enables their proliferation. Two MEKis, selumetinib and trametinib, induce T cell activation with increased CTLA-4 expression and, to a lesser extent, PD-1 expression on T cells in vivo after cyclical pulsatile MEKi treatment. In addition, the pulsatile dosing schedule alone shows superior anti-tumor effects and delays the emergence of drug resistance. Furthermore, pulsatile MEKi treatment combined with CTLA-4 blockade prolongs survival in mice bearing tumors with mutant Kras. Our results set the foundation and show the importance of a combinatorial therapeutic strategy using pulsatile targeted therapy together with immunotherapy to optimally enhance tumor delay and promote long-term anti-tumor immunity.
Copyright © 2019 The Authors. Published by Elsevier Inc. All rights reserved.
Conflict of interest statement
DECLARATION OF INTERESTS
P.D.S. is an employee and shareholder of AstraZeneca.
T.M. is a cofounder and holds an equity in IMVAQ Therapeutics. He is a consultant of Immunos Therapeutics and Pfizer. He has research support from Bristol-Myers Squibb; Surface Oncology; Kyn Therapeutics; Infinity Pharmaceuticals, Inc.; Peregrine Pharmeceuticals, Inc.; Adaptive Biotechnologies; Leap Therapeutics, Inc.; and Aprea. He has patents on applications related to work on oncolytic viral therapy, alpha virus-based vaccine, neo antigen modeling, CD40, GITR, OX40, PD-1, and CTLA-4.
K.K.W. is a founder and equity holder of G1 Therapeutics. He has sponsored research agreements with MedImmune, Takeda, TargImmune, and BMS. He also has consulting and sponsored research agreements with AstraZeneca, Janssen, Pfizer, Novartis, Merck, Ono, and Array.
J.D.W. is a consultant for Adaptive Biotech; Advaxis; Amgen; Apricity; Array BioPharma; Ascentage Pharma; Astellas; Bayer; Beigene; Bristol Myers Squibb; Celgene; Chugai; Elucida; Eli Lilly; F Star; Genentech; Imvaq; Janssen; Kleo Pharma; Linneaus; MedImmune; Merck; Neon Therapuetics; Ono; Polaris Pharma; Polynoma; Psioxus; Puretech; Recepta; Trieza; Sellas Life Sciences; Serametrix; Surface Oncology; and Syndax. He has research support from Bristol Myers Squibb, Medimmune, and Genentech. He holds equity in Potenza Therapeutics; Tizona Pharmaceuticals; Adaptive Biotechnologies; Elu-cida; Imvaq; Beigene; Trieza; and Linneaus and has an honorarium from Esanex. He has patents of xenogeneic DNA vaccines (royalties); alphavirus replicon particles expressing TRP2; myeloid-derived suppressor cell (MDSC) assay (royalties); Newcastle disease viruses for cancer therapy; a genomic signature to identify responders to ipilimumab in melanoma; engineered vaccinia viruses for cancer immunotherapy; an anti-CD40 agonist monoclonal antibody (mAb) fused to monophosphoryl lipid A (MPL) for cancer therapy; CAR T cells targeting differentiation antigens as means to treat cancer; an anti-PD1 antibody; anti-CTLA4 antibodies; and anti-GITR antibodies and methods of use thereof.
The other authors declare no competing interests.
Figures
Similar articles
-
The Combination of MEK Inhibitor With Immunomodulatory Antibodies Targeting Programmed Death 1 and Programmed Death Ligand 1 Results in Prolonged Survival in Kras/p53-Driven Lung Cancer.J Thorac Oncol. 2019 Jun;14(6):1046-1060. doi: 10.1016/j.jtho.2019.02.004. Epub 2019 Feb 13. J Thorac Oncol. 2019. PMID: 30771521 Free PMC article.
-
Enhancing Therapeutic Efficacy of Oncolytic Herpes Simplex Virus with MEK Inhibitor Trametinib in Some BRAF or KRAS-Mutated Colorectal or Lung Carcinoma Models.Viruses. 2021 Sep 3;13(9):1758. doi: 10.3390/v13091758. Viruses. 2021. PMID: 34578339 Free PMC article.
-
Th17 cells contribute to combination MEK inhibitor and anti-PD-L1 therapy resistance in KRAS/p53 mutant lung cancers.Nat Commun. 2021 May 10;12(1):2606. doi: 10.1038/s41467-021-22875-w. Nat Commun. 2021. PMID: 33972557 Free PMC article.
-
Targeting of MEK in lung cancer therapeutics.Lancet Respir Med. 2015 Apr;3(4):319-27. doi: 10.1016/S2213-2600(15)00026-0. Epub 2015 Mar 20. Lancet Respir Med. 2015. PMID: 25801412 Review.
-
Therapeutic potential of trametinib to inhibit the mutagenesis by inactivating the protein kinase pathway in non-small cell lung cancer.Expert Rev Anticancer Ther. 2019 Jan;19(1):11-17. doi: 10.1080/14737140.2019.1554440. Epub 2018 Dec 4. Expert Rev Anticancer Ther. 2019. PMID: 30513023 Review.
Cited by
-
Precision Nutrition and Cancer Relapse Prevention: A Systematic Literature Review.Nutrients. 2019 Nov 16;11(11):2799. doi: 10.3390/nu11112799. Nutrients. 2019. PMID: 31744117 Free PMC article.
-
New Horizons in KRAS-Mutant Lung Cancer: Dawn After Darkness.Front Oncol. 2019 Sep 25;9:953. doi: 10.3389/fonc.2019.00953. eCollection 2019. Front Oncol. 2019. PMID: 31612108 Free PMC article. Review.
-
Relationship between Lung Carcinogenesis and Chronic Inflammation in Rodents.Cancers (Basel). 2021 Jun 10;13(12):2910. doi: 10.3390/cancers13122910. Cancers (Basel). 2021. PMID: 34200786 Free PMC article. Review.
-
Targeted inhibition of glutamine metabolism enhances the antitumor effect of selumetinib in KRAS-mutant NSCLC.Transl Oncol. 2021 Jan;14(1):100920. doi: 10.1016/j.tranon.2020.100920. Epub 2020 Nov 1. Transl Oncol. 2021. PMID: 33137541 Free PMC article.
-
Targeting KRAS(G12C): From Inhibitory Mechanism to Modulation of Antitumor Effects in Patients.Cell. 2020 Nov 12;183(4):850-859. doi: 10.1016/j.cell.2020.09.044. Epub 2020 Oct 15. Cell. 2020. PMID: 33065029 Free PMC article. Review.
References
-
- Andrzejewski S, Klimcakova E, Johnson RM, Tabariés S, Annis MG, McGuirk S, Northey JJ, Chénard V, Sriram U, Papadopoli DJ, et al. (2017). PGC-1a promotes breast cancer metastasis and confers bioenergetic flexibility against metabolic drugs. Cell Metab 26, 778–787.e5. - PubMed
-
- Bolstad BM, Irizarry RA, Astrand M, and Speed TP (2003). A comparison of normalization methods for high density oligonucleotide array data based on variance and bias. Bioinformatics 19, 185–193. - PubMed
-
- Brant R, Sharpe A, Liptrot T, Dry JR, Harrington EA, Barrett JC, Whalley N, Womack C, Smith P, and Hodgson DR (2017). Clinically viable gene expression assays with potential for predicting benefit from MEK inhibitors. Clin. Cancer Res 23, 1471–1480. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Miscellaneous
