

Immune Signaling in Neurodegeneration
- PMID: 30995509
- PMCID: PMC6822103
- DOI: 10.1016/j.immuni.2019.03.016
Immune Signaling in Neurodegeneration
Abstract
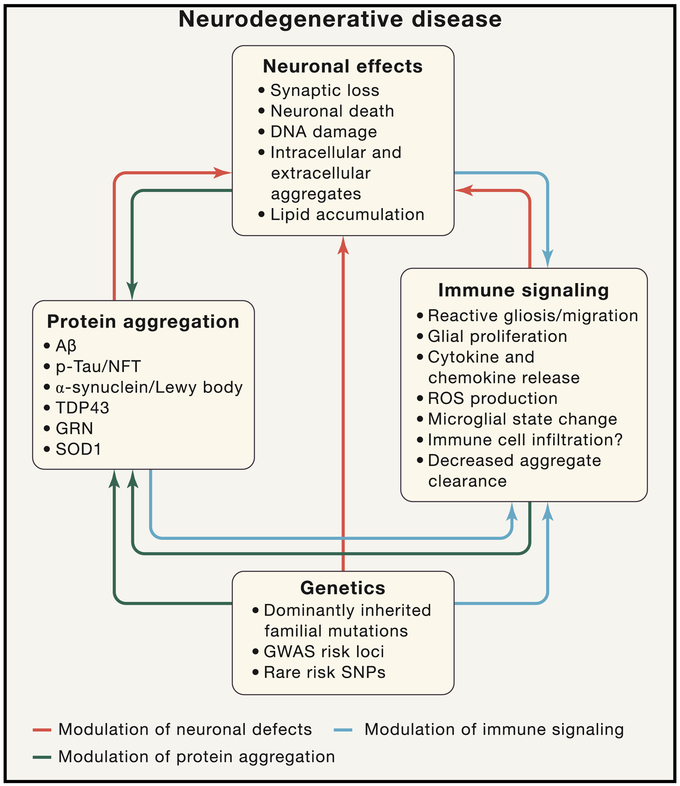
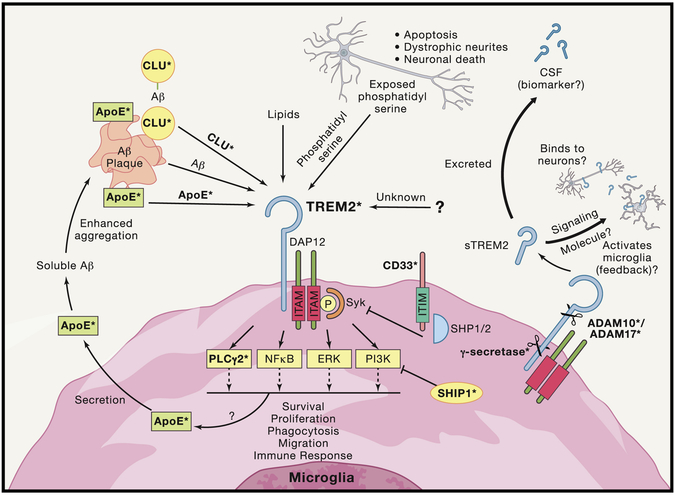
Neurodegenerative diseases of the central nervous system progressively rob patients of their memory, motor function, and ability to perform daily tasks. Advances in genetics and animal models are beginning to unearth an unexpected role of the immune system in disease onset and pathogenesis; however, the role of cytokines, growth factors, and other immune signaling pathways in disease pathogenesis is still being examined. Here we review recent genetic risk and genome-wide association studies and emerging mechanisms for three key immune pathways implicated in disease, the growth factor TGF-β, the complement cascade, and the extracellular receptor TREM2. These immune signaling pathways are important under both healthy and neurodegenerative conditions, and recent work has highlighted new functional aspects of their signaling. Finally, we assess future directions for immune-related research in neurodegeneration and potential avenues for immune-related therapies.
Keywords: Alzheimer’s disease; Complement; Cytokine; GWAS; Inflammation; Innate Immunity; Microglia; Neuro-immune; Neurodegeneration; Neuroinflammation; Signaling; TGF-β; TREM2.
Copyright © 2019 Elsevier Inc. All rights reserved.
Conflict of interest statement
Conflicts of Interest:
No conflicts of interest to declare.
B.S. is a member of the Scientific Advisory Board and minor shareholder in Annexon Biosciences. B.S. is an inventor on multiple patents: “Modulation of Synaptic Maintenance” (US8148330B2, US9149444B2) and is inventor on pending patent “Biomarkers for dementia and dementia related neurological disorders” (WO2015103594A1/US20160327572A1). T.R.H. & S.E.M. have no interests to declare.
Figures


References
-
- Afagh A, Cummings BJ, Cribbs DH, Cotman CW, and Tenner AJ (1996). Localization and cell association of C1q in Alzheimer's disease brain. Exp Neurol 138, 22–32. - PubMed
-
- Alzheimer A (1907). Über eine eigenartige Erkrankung der Hirnrinde. Allgemeine Zeitschrift für Psychiatrie und psychisch-gerichtliche Medizin 64.
-
- Alzheimer A (1911). Über eigenartige Krankheitsfälle des späteren Alters.
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
