

Novel, rare and common pathogenic variants in the CFTR gene screened by high-throughput sequencing technology and predicted by in silico tools
- PMID: 30996306
- PMCID: PMC6470152
- DOI: 10.1038/s41598-019-42404-6
Novel, rare and common pathogenic variants in the CFTR gene screened by high-throughput sequencing technology and predicted by in silico tools
Abstract
Cystic fibrosis (CF) is caused by ~300 pathogenic CFTR variants. The heterogeneity of which, challenges molecular diagnosis and precision medicine approaches in CF. Our objective was to identify CFTR variants through high-throughput sequencing (HTS) and to predict the pathogenicity of novel variants through in 8 silico tools. Two guidelines were followed to deduce the pathogenicity. A total of 169 CF patients had genomic DNA submitted to a Targeted Gene Sequencing and we identified 63 variants (three patients had three variants). The most frequent alleles were: F508del (n = 192), G542* (n = 26), N1303K (n = 11), R1162* and R334W (n = 9). The screened variants were classified as follows: 41 - pathogenic variants [classified as (I) n = 23, (II) n = 6, (III) n = 1, (IV) n = 6, (IV/V) n = 1 and (VI) n = 4]; 14 - variants of uncertain significance; and seven novel variants. To the novel variants we suggested the classification of 6b-16 exon duplication, G646* and 3557delA as Class I. There was concordance among the predictors as likely pathogenic for L935Q, cDNA.5808T>A and I1427I. Also, Y325F presented two discordant results among the predictors. HTS and in silico analysis can identify pathogenic CFTR variants and will open the door to integration of precision medicine into routine clinical practice in the near future.
Conflict of interest statement
FALM: Fundação de Amparo à Pesquisa do Estado de São Paulo (FAPESP) for the research grants and scholarships #2011/12939-4, #2011/18845-1, #2015/12183-8 and #2015/12858-5; Fundo de Apoio à Pesquisa, ao Ensino e à Extensão da Universidade Estadual de Campinas for supporting the research #0648/2015; JDR: FAPESP for assisting the research 2011/18845-1 and #2015/12183-8. Also, we thank VERTEX for providing us with the financial support to screen the
Figures
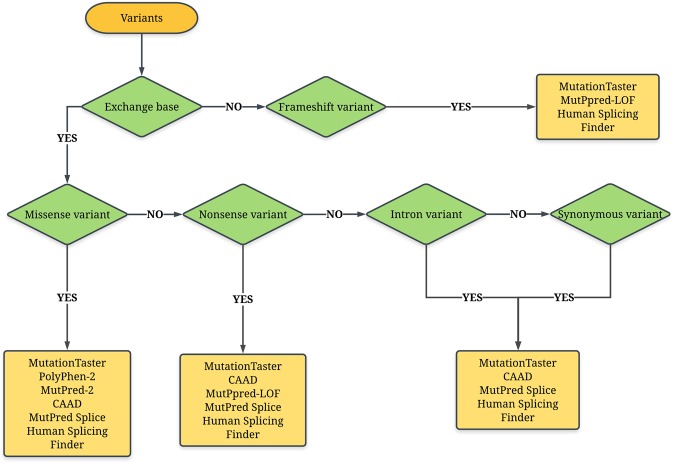
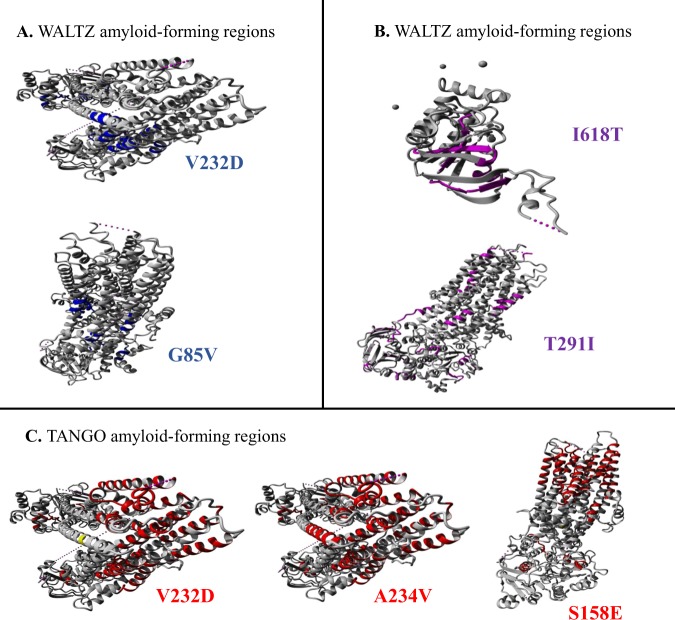
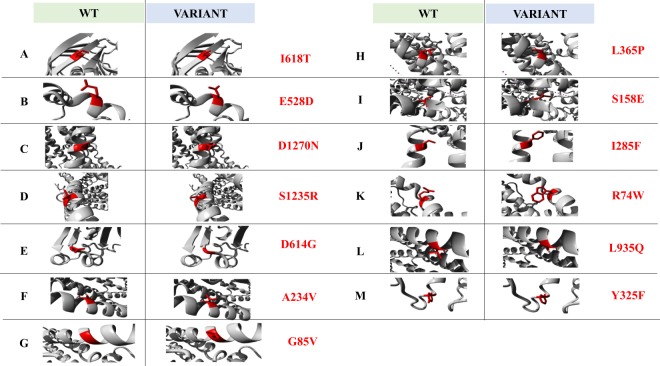
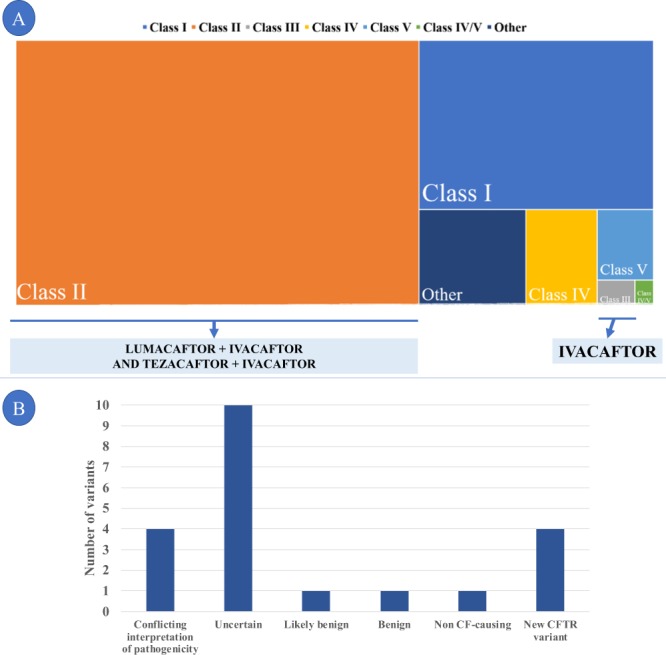
References
-
- Cystic Fibrosis Mutation Database, 2018, http://www. genet.sickkids.on.ca/Home.html (Accessed June 2018).
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
