

Sequence imputation from low density single nucleotide polymorphism panel in a black poplar breeding population
- PMID: 30999856
- PMCID: PMC6471894
- DOI: 10.1186/s12864-019-5660-y
Sequence imputation from low density single nucleotide polymorphism panel in a black poplar breeding population
Abstract
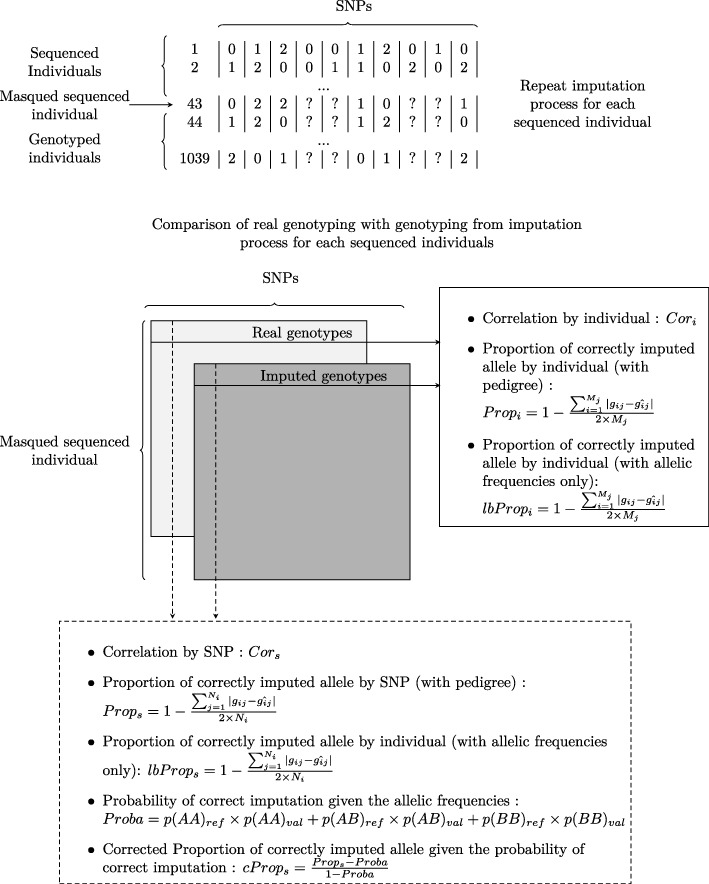
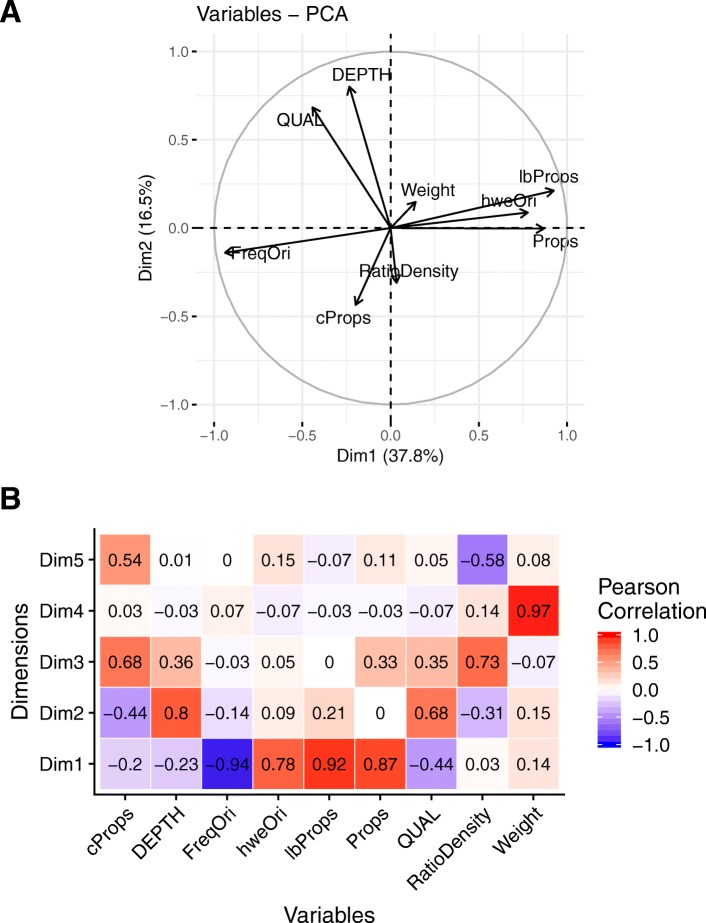
Background: Genomic selection accuracy increases with the use of high SNP (single nucleotide polymorphism) coverage. However, such gains in coverage come at high costs, preventing their prompt operational implementation by breeders. Low density panels imputed to higher densities offer a cheaper alternative during the first stages of genomic resources development. Our study is the first to explore the imputation in a tree species: black poplar. About 1000 pure-breed Populus nigra trees from a breeding population were selected and genotyped with a 12K custom Infinium Bead-Chip. Forty-three of those individuals corresponding to nodal trees in the pedigree were fully sequenced (reference), while the remaining majority (target) was imputed from 8K to 1.4 million SNPs using FImpute. Each SNP and individual was evaluated for imputation errors by leave-one-out cross validation in the training sample of 43 sequenced trees. Some summary statistics such as Hardy-Weinberg Equilibrium exact test p-value, quality of sequencing, depth of sequencing per site and per individual, minor allele frequency, marker density ratio or SNP information redundancy were calculated. Principal component and Boruta analyses were used on all these parameters to rank the factors affecting the quality of imputation. Additionally, we characterize the impact of the relatedness between reference population and target population.
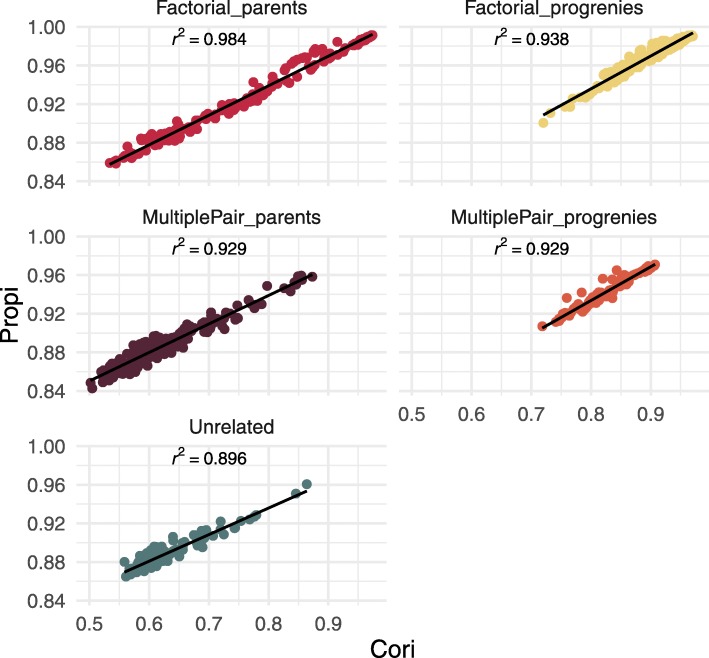
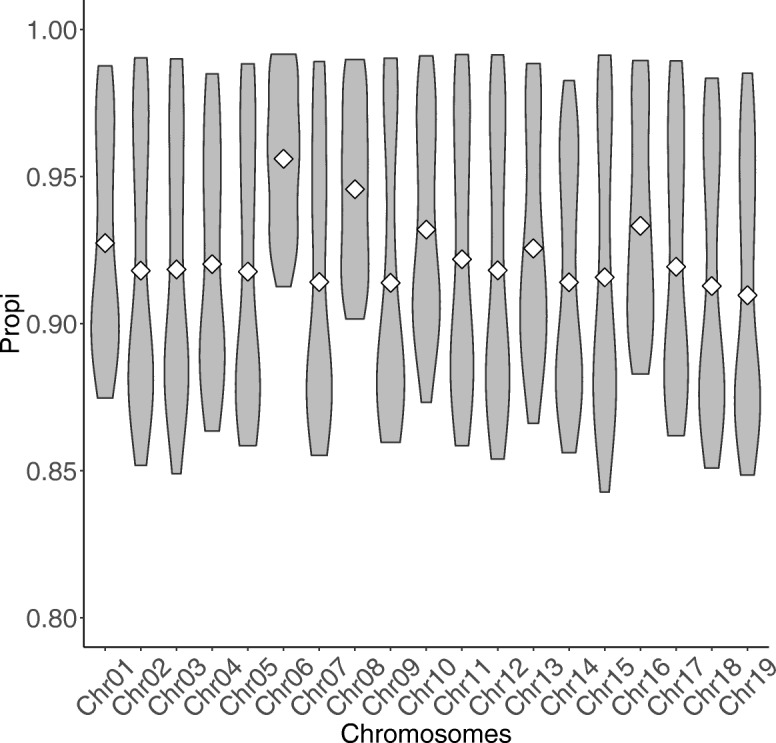
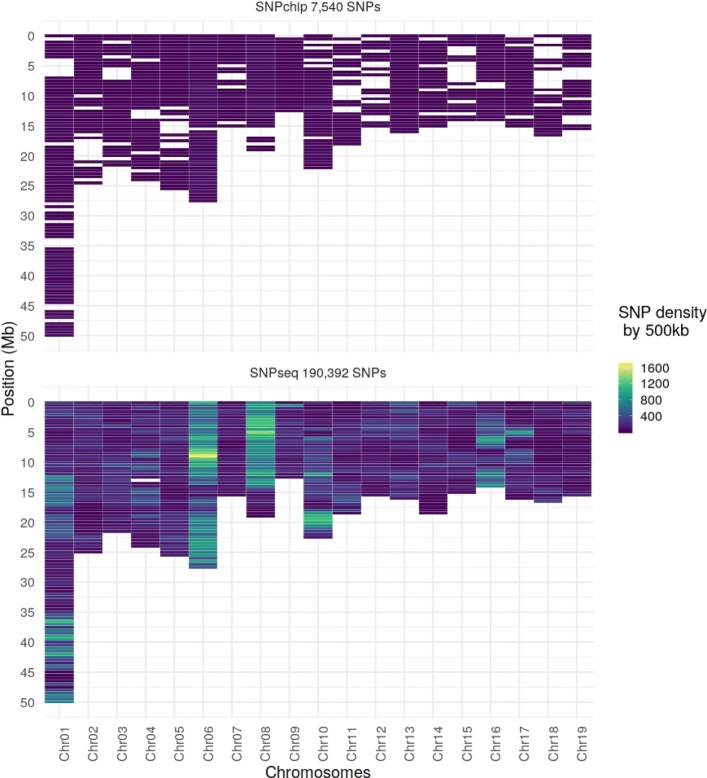
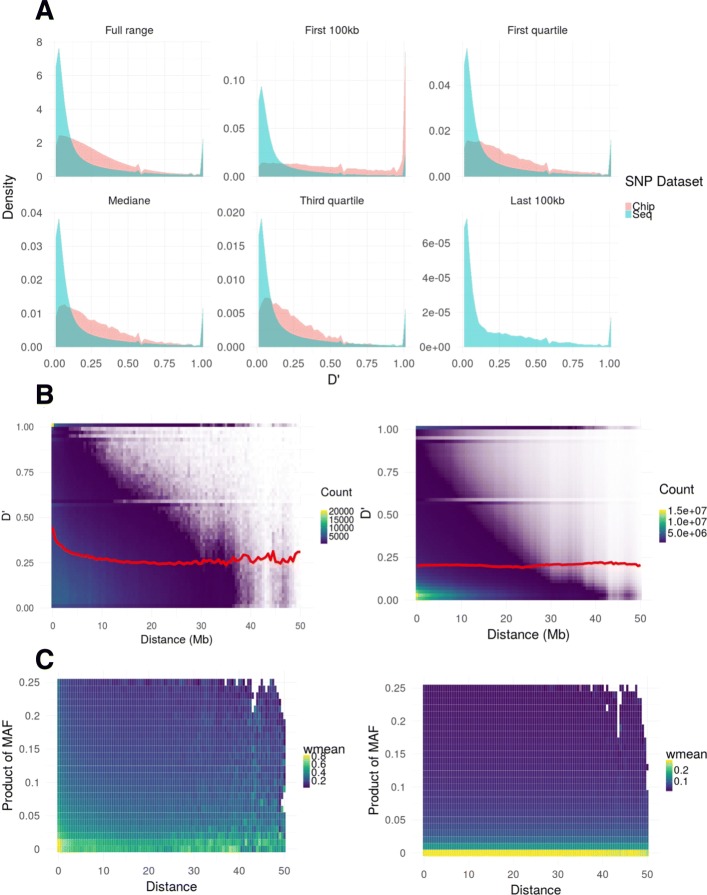
Results: During the imputation process, we used 7540 SNPs from the chip to impute 1,438,827 SNPs from sequences. At the individual level, imputation accuracy was high with a proportion of SNPs correctly imputed between 0.84 and 0.99. The variation in accuracies was mostly due to differences in relatedness between individuals. At a SNP level, the imputation quality depended on genotyped SNP density and on the original minor allele frequency. The imputation did not appear to result in an increase of linkage disequilibrium. The genotype densification not only brought a better distribution of markers all along the genome, but also we did not detect any substantial bias in annotation categories.
Conclusions: This study shows that it is possible to impute low-density marker panels to whole genome sequence with good accuracy under certain conditions that could be common to many breeding populations.
Keywords: Genotype Imputation; Low density arrays; Populus nigra; Whole-Genome Resequencing.
Conflict of interest statement
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Figures
References
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
