

Forging tools for refining predicted protein structures
- PMID: 31000596
- PMCID: PMC6511001
- DOI: 10.1073/pnas.1900778116
Forging tools for refining predicted protein structures
Abstract
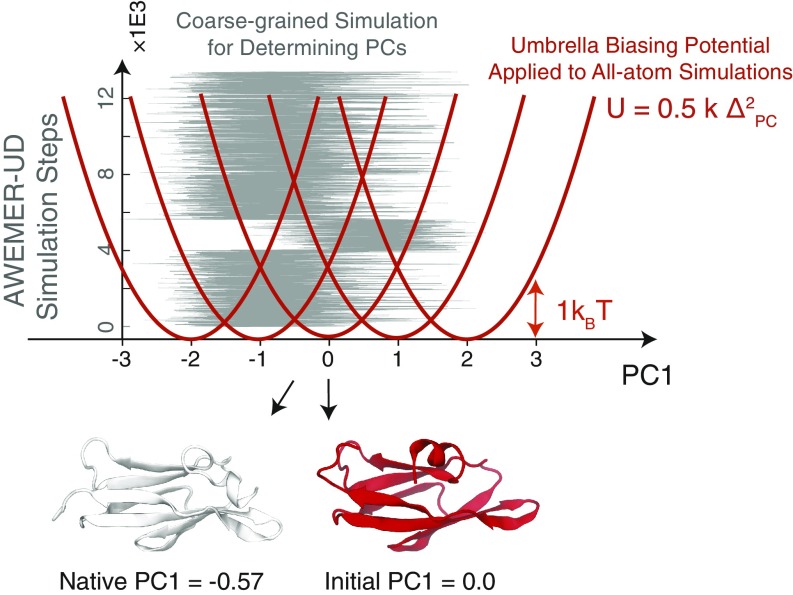
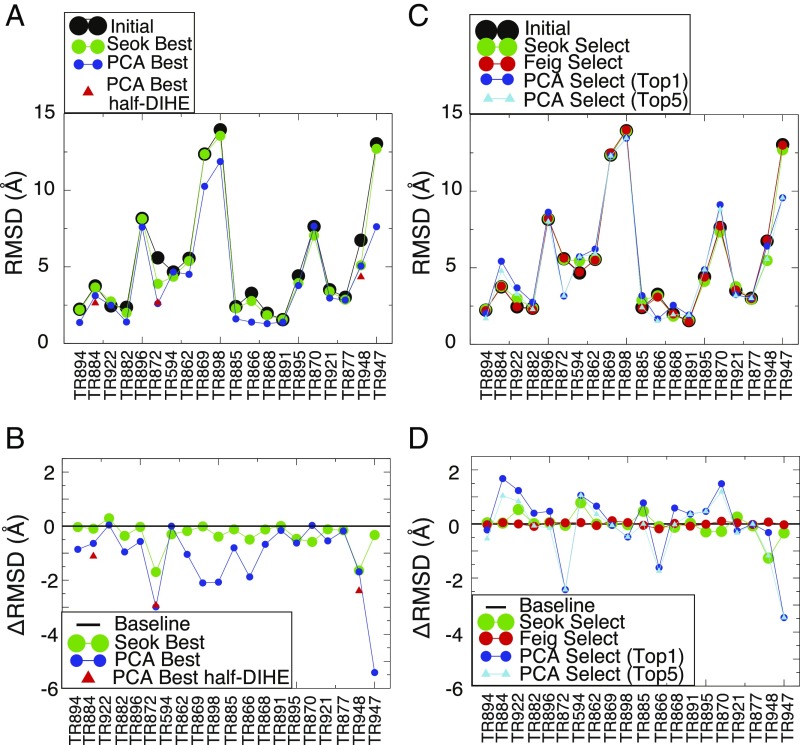
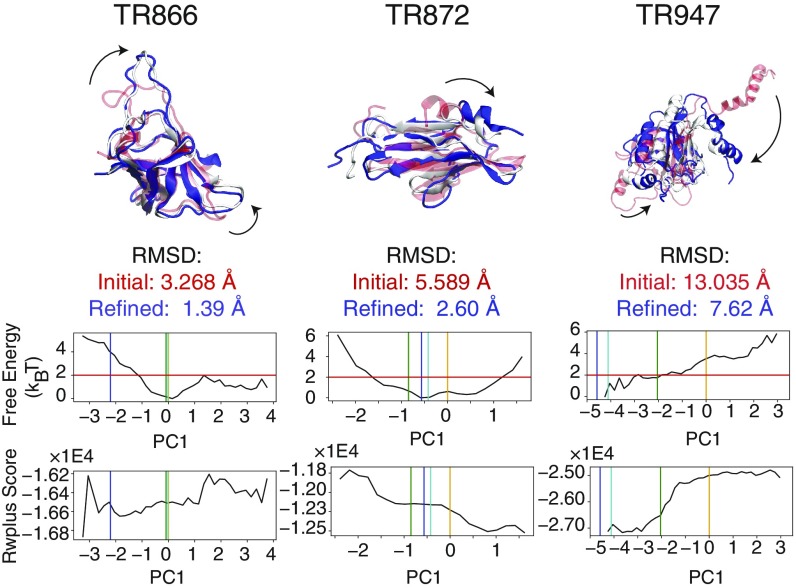
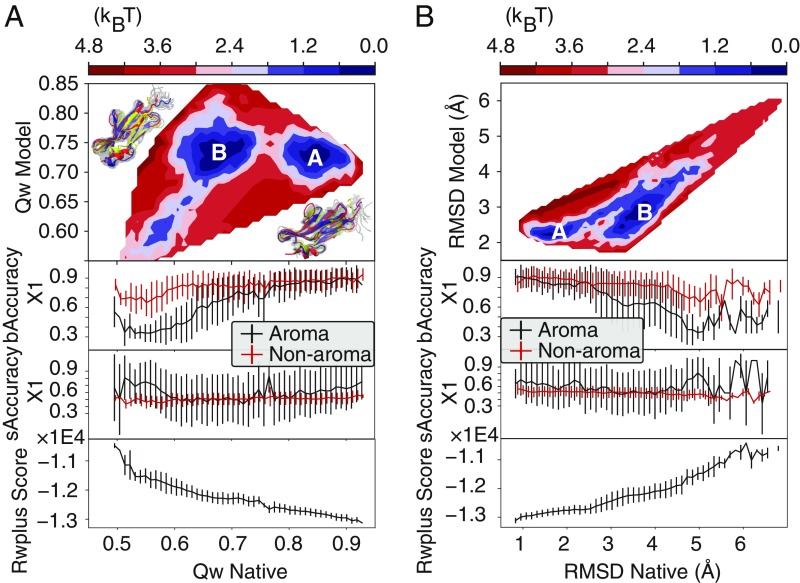
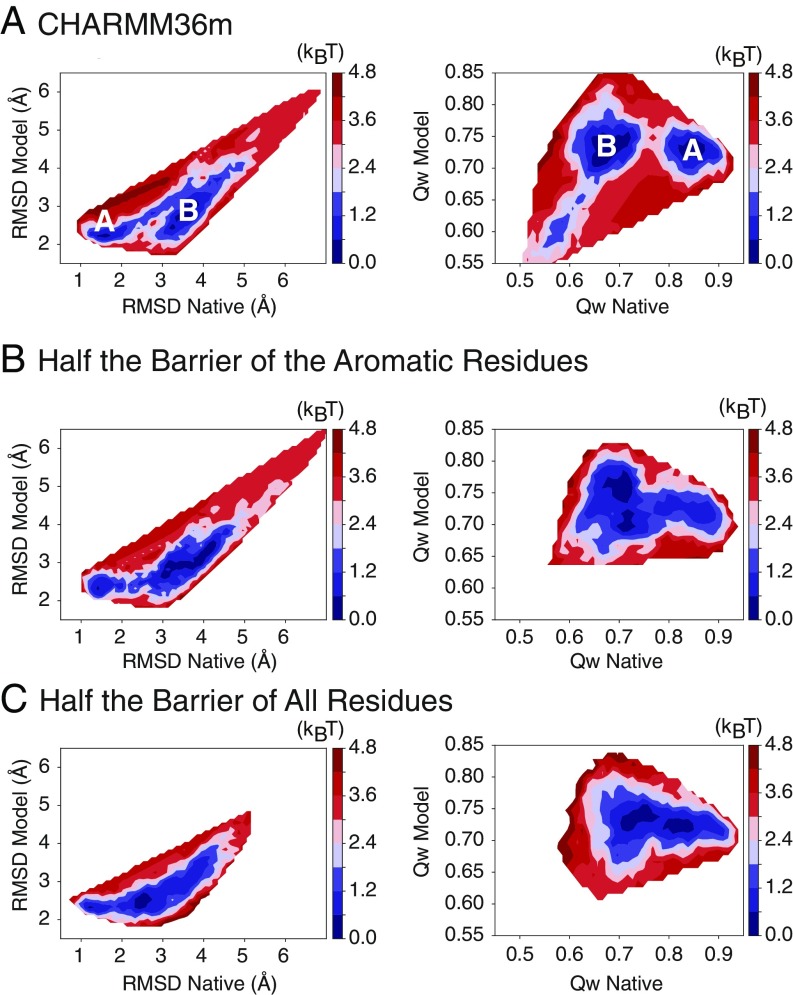
Refining predicted protein structures with all-atom molecular dynamics simulations is one route to producing, entirely by computational means, structural models of proteins that rival in quality those that are determined by X-ray diffraction experiments. Slow rearrangements within the compact folded state, however, make routine refinement of predicted structures by unrestrained simulations infeasible. In this work, we draw inspiration from the fields of metallurgy and blacksmithing, where practitioners have worked out practical means of controlling equilibration by mechanically deforming their samples. We describe a two-step refinement procedure that involves identifying collective variables for mechanical deformations using a coarse-grained model and then sampling along these deformation modes in all-atom simulations. Identifying those low-frequency collective modes that change the contact map the most proves to be an effective strategy for choosing which deformations to use for sampling. The method is tested on 20 refinement targets from the CASP12 competition and is found to induce large structural rearrangements that drive the structures closer to the experimentally determined structures during relatively short all-atom simulations of 50 ns. By examining the accuracy of side-chain rotamer states in subensembles of structures that have varying degrees of similarity to the experimental structure, we identified the reorientation of aromatic side chains as a step that remains slow even when encouraging global mechanical deformations in the all-atom simulations. Reducing the side-chain rotamer isomerization barriers in the all-atom force field is found to further speed up refinement.
Keywords: mechanical deformations; principal component analysis; protein blacksmithing; protein structure refinement; side-chain isomerization.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous
