

Modulation of Allergic Inflammation in the Nasal Mucosa of Allergic Rhinitis Sufferers With Topical Pharmaceutical Agents
- PMID: 31001114
- PMCID: PMC6455085
- DOI: 10.3389/fphar.2019.00294
Modulation of Allergic Inflammation in the Nasal Mucosa of Allergic Rhinitis Sufferers With Topical Pharmaceutical Agents
Abstract
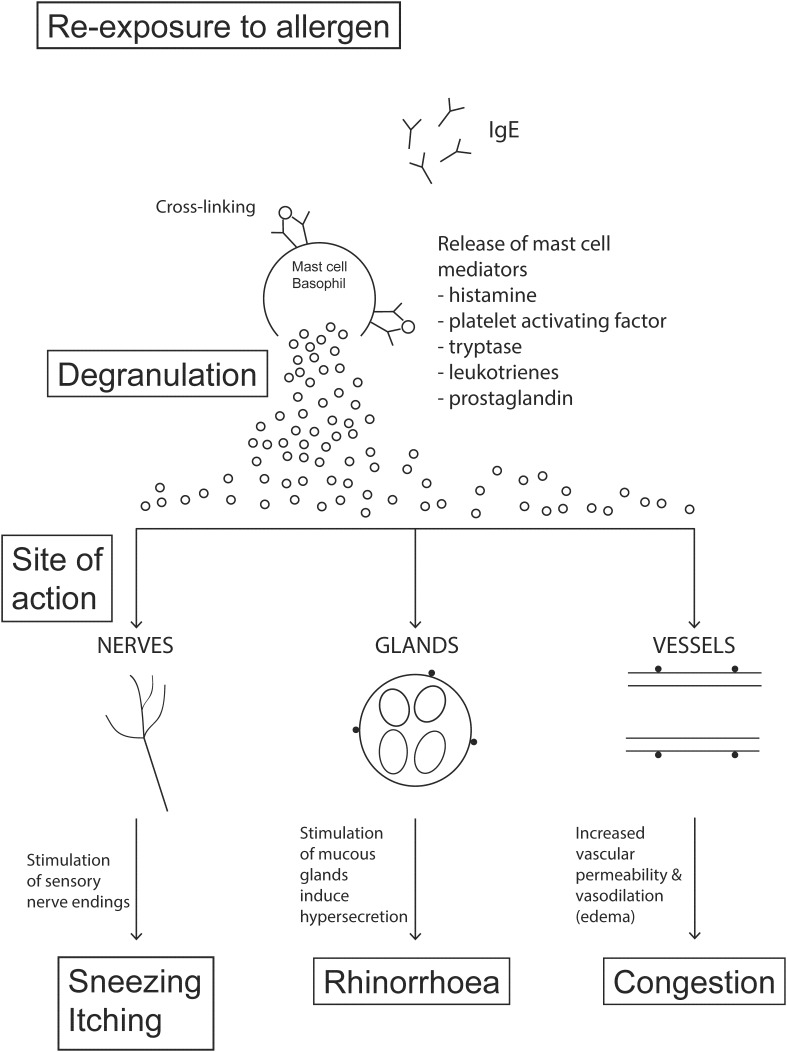
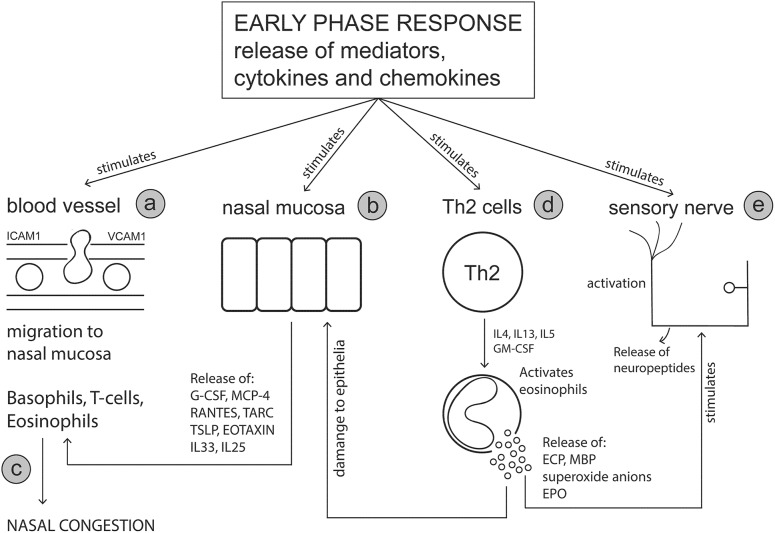
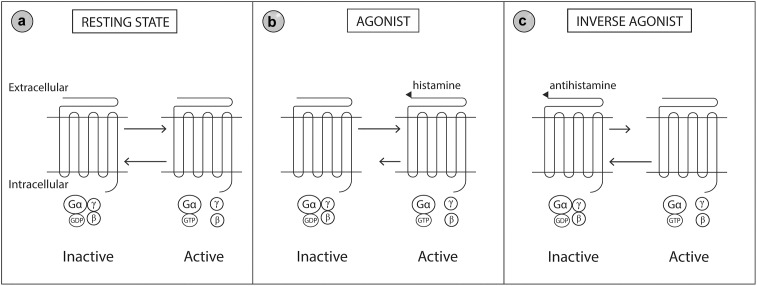
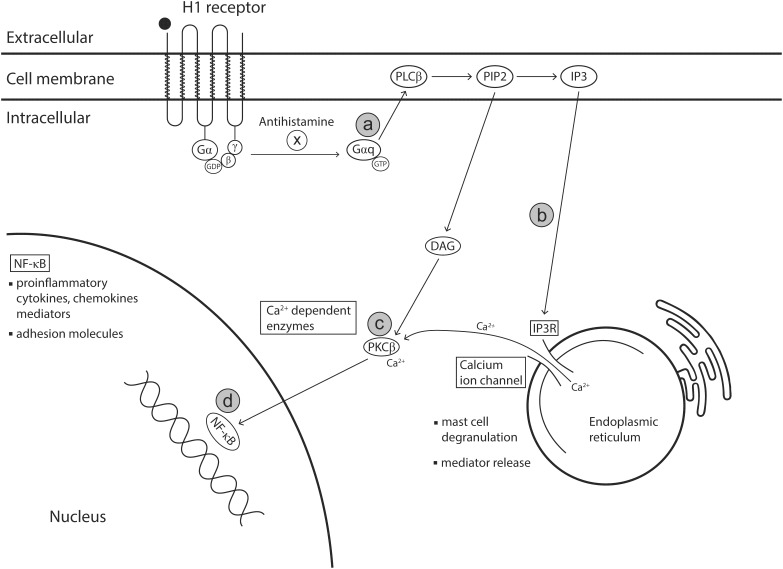
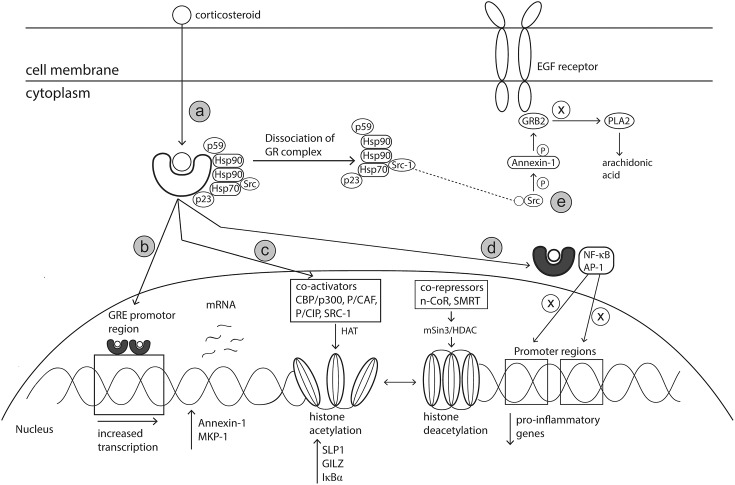
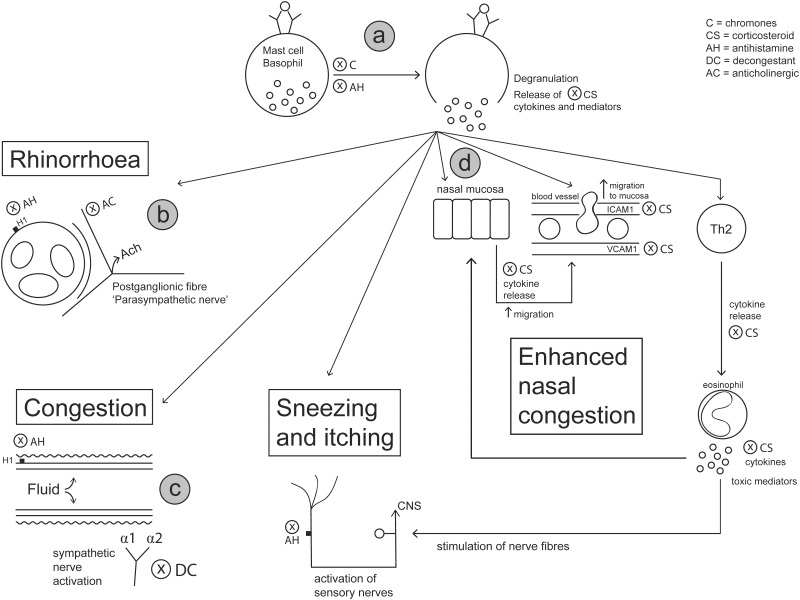
Allergic rhinitis (AR) is a chronic upper respiratory disease estimated to affect between 10 and 40% of the worldwide population. The mechanisms underlying AR are highly complex and involve multiple immune cells, mediators, and cytokines. As such, the development of a single drug to treat allergic inflammation and/or symptoms is confounded by the complexity of the disease pathophysiology. Complete avoidance of allergens that trigger AR symptoms is not possible and without a cure, the available therapeutic options are typically focused on achieving symptomatic relief. Topical therapies offer many advantages over oral therapies, such as delivering greater concentrations of drugs to the receptor sites at the source of the allergic inflammation and the reduced risk of systemic side effects. This review describes the complex pathophysiology of AR and identifies the mechanism(s) of action of topical treatments including antihistamines, steroids, anticholinergics, decongestants and chromones in relation to AR pathophysiology. Following the literature review a discussion on the future therapeutic strategies for AR treatment is provided.
Keywords: allergic rhinitis; anticholinergic; antihistamines; chromones; decongestants; intranasal; steroids.
Figures
References
Publication types
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials