

Diverse Large HIV-1 Non-subtype B Clusters Are Spreading Among Men Who Have Sex With Men in Spain
- PMID: 31001231
- PMCID: PMC6457325
- DOI: 10.3389/fmicb.2019.00655
Diverse Large HIV-1 Non-subtype B Clusters Are Spreading Among Men Who Have Sex With Men in Spain
Abstract
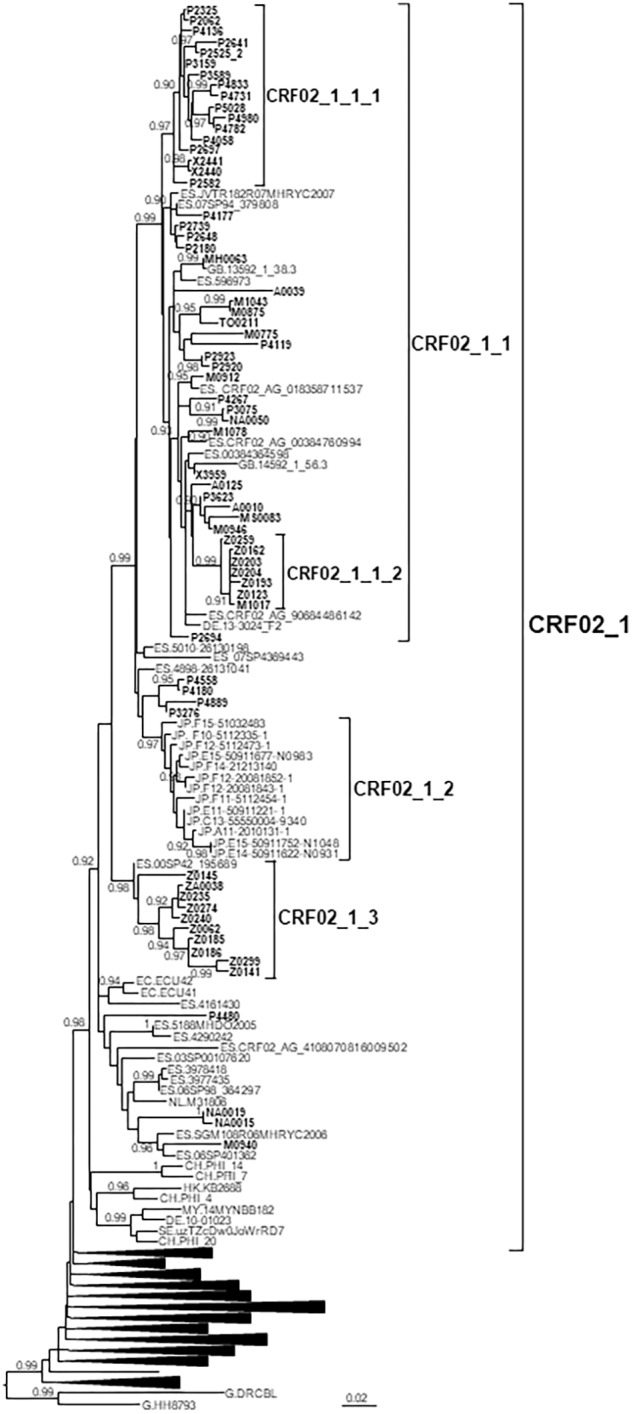
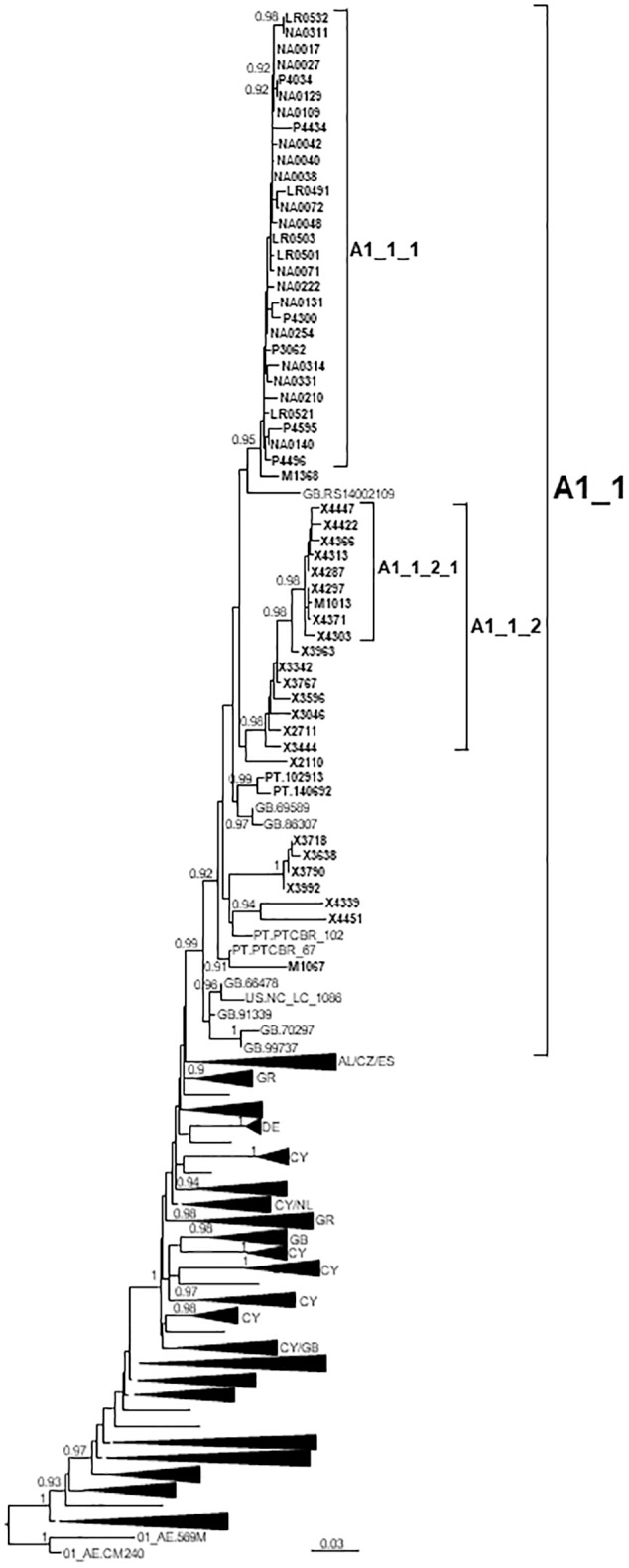
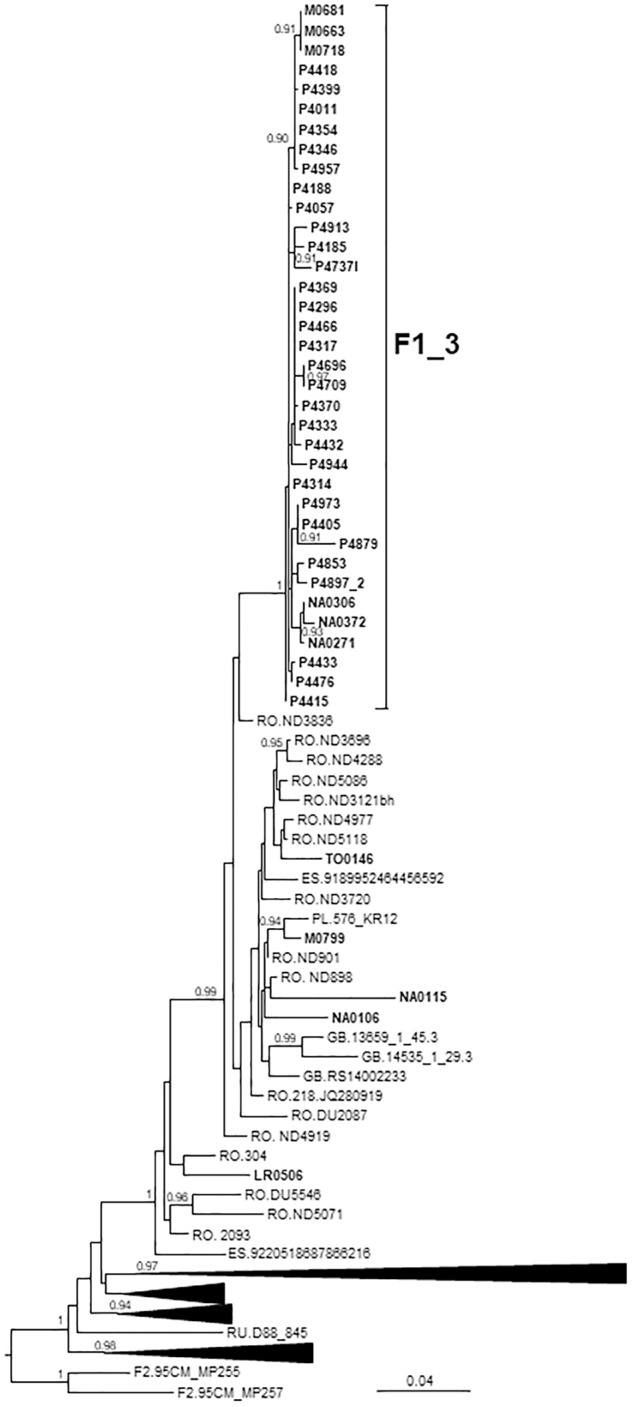
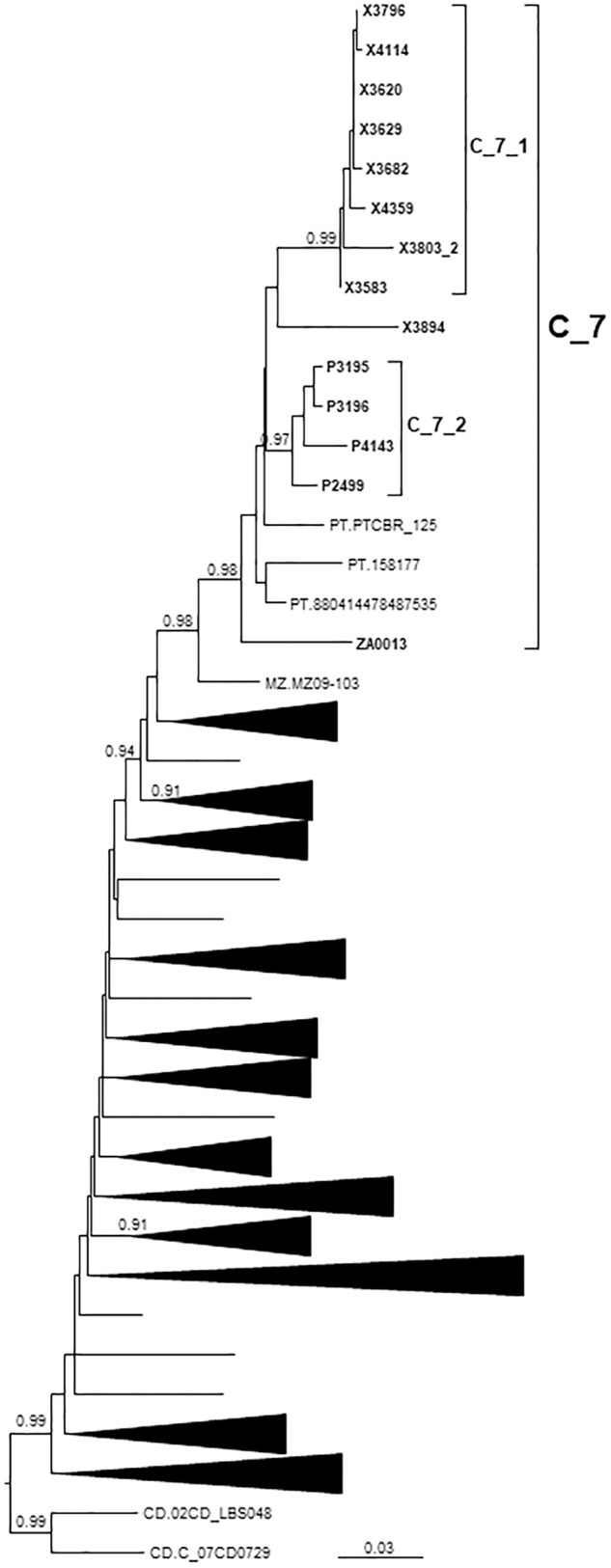
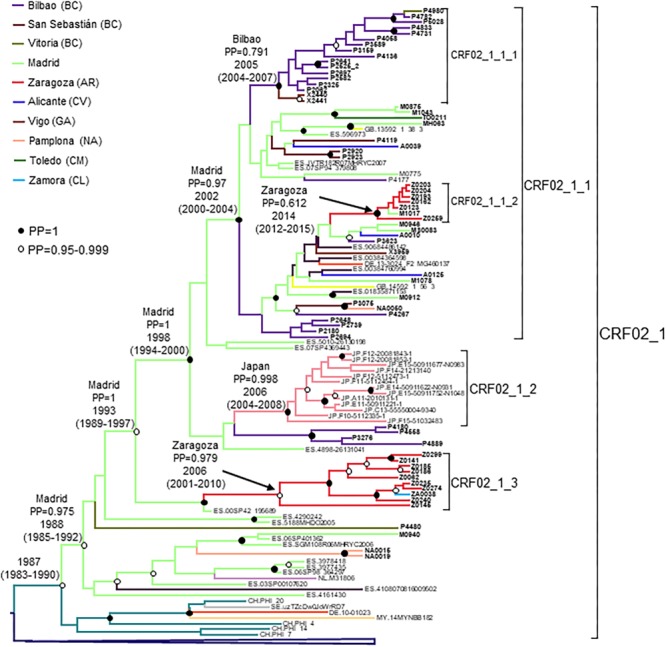
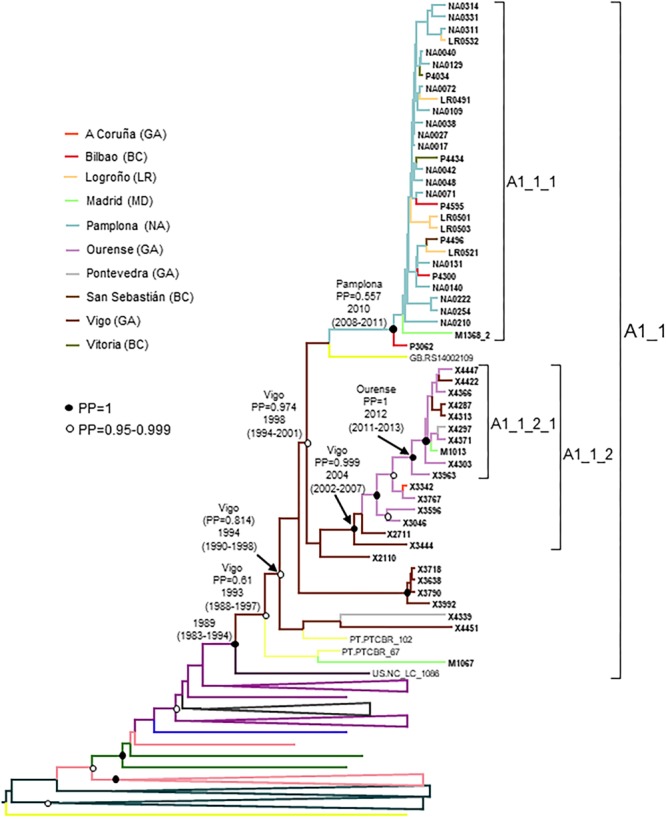
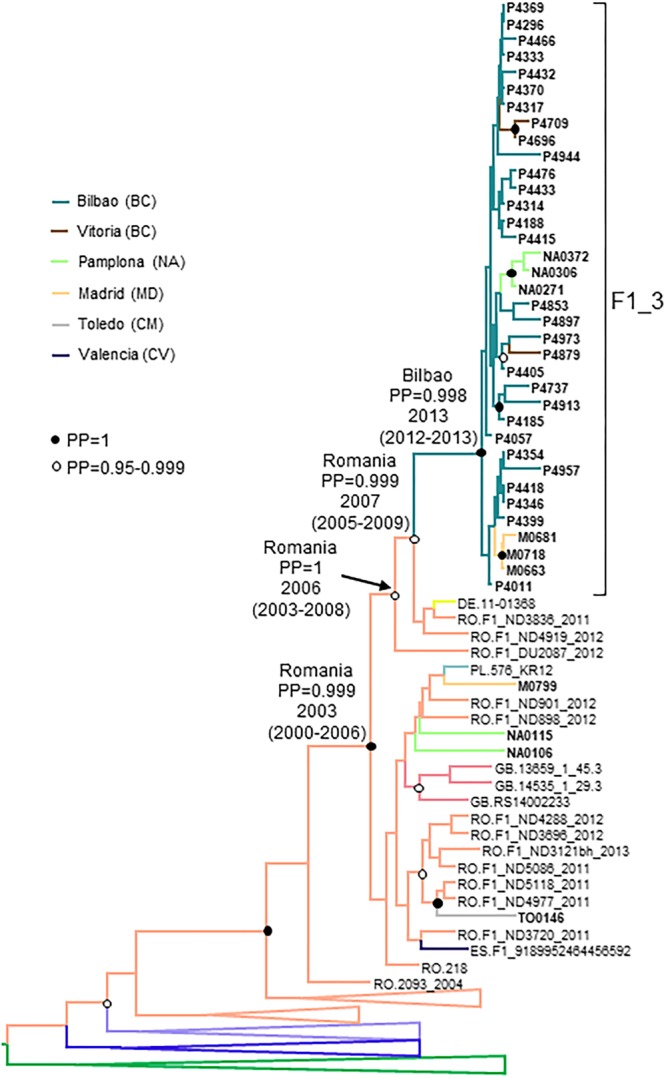
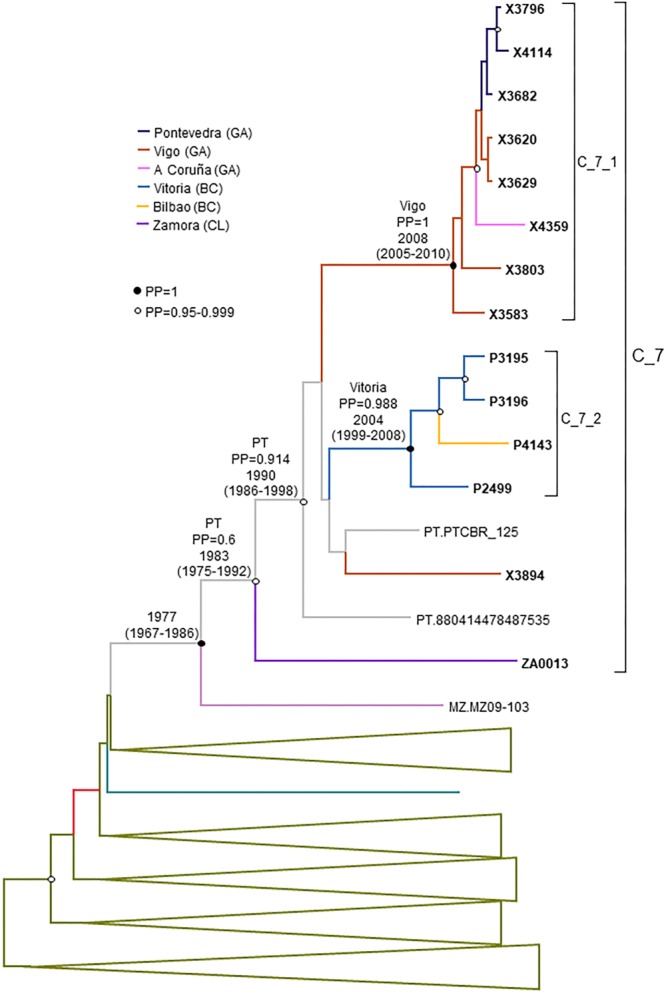
In Western Europe, the HIV-1 epidemic among men who have sex with men (MSM) is dominated by subtype B. However, recently, other genetic forms have been reported to circulate in this population, as evidenced by their grouping in clusters predominantly comprising European individuals. Here we describe four large HIV-1 non-subtype B clusters spreading among MSM in Spain. Samples were collected in 9 regions. A pol fragment was amplified from plasma RNA or blood-extracted DNA. Phylogenetic analyses were performed via maximum likelihood, including database sequences of the same genetic forms as the identified clusters. Times and locations of the most recent common ancestors (MRCA) of clusters were estimated with a Bayesian method. Five large non-subtype B clusters associated with MSM were identified. The largest one, of F1 subtype, was reported previously. The other four were of CRF02_AG (CRF02_1; n = 115) and subtypes A1 (A1_1; n = 66), F1 (F1_3; n = 36), and C (C_7; n = 17). Most individuals belonging to them had been diagnosed of HIV-1 infection in the last 10 years. Each cluster comprised viruses from 3 to 8 Spanish regions and also comprised or was related to viruses from other countries: CRF02_1 comprised a Japanese subcluster and viruses from 8 other countries from Western Europe, Asia, and South America; A1_1 comprised viruses from Portugal, United Kingom, and United States, and was related to the A1 strain circulating in Greece, Albania and Cyprus; F1_3 was related to viruses from Romania; and C_7 comprised viruses from Portugal and was related to a virus from Mozambique. A subcluster within CRF02_1 was associated with heterosexual transmission. Near full-length genomes of each cluster were of uniform genetic form. Times of MRCAs of CRF02_1, A1_1, F1_3, and C_7 were estimated around 1986, 1989, 2013, and 1983, respectively. MRCA locations for CRF02_1 and A1_1 were uncertain (however initial expansions in Spain in Madrid and Vigo, respectively, were estimated) and were most probable in Bilbao, Spain, for F1_3 and Portugal for C_7. These results show that the HIV-1 epidemic among MSM in Spain is becoming increasingly diverse through the expansion of diverse non-subtype B clusters, comprising or related to viruses circulating in other countries.
Keywords: HIV-1; circulating recombinant forms; clusters; men who have sex with men; molecular epidemiology; phylodynamics; phylogeny; subtypes.
Figures
Similar articles
-
Phylogeny and Phylogeography of a Recent HIV-1 Subtype F Outbreak among Men Who Have Sex with Men in Spain Deriving from a Cluster with a Wide Geographic Circulation in Western Europe.PLoS One. 2015 Nov 24;10(11):e0143325. doi: 10.1371/journal.pone.0143325. eCollection 2015. PLoS One. 2015. PMID: 26599410 Free PMC article.
-
HIV-1 transmission networks across Cyprus (2010-2012).PLoS One. 2018 Apr 23;13(4):e0195660. doi: 10.1371/journal.pone.0195660. eCollection 2018. PLoS One. 2018. PMID: 29684083 Free PMC article.
-
[HIV-1 subtype distribution determined by phylogenetic analysis of pol gene sequences and automated subtyping tools among HIV-1 isolates from the Aegian Region of Turkey].Mikrobiyol Bul. 2014 Jul;48(3):420-8. doi: 10.5578/mb.7609. Mikrobiyol Bul. 2014. PMID: 25052108 Turkish.
-
Molecular epidemiology of HIV-1 infection in Europe: An overview.Infect Genet Evol. 2016 Dec;46:180-189. doi: 10.1016/j.meegid.2016.06.033. Epub 2016 Jun 16. Infect Genet Evol. 2016. PMID: 27321440 Review.
-
Romania, a Harbour of HIV-1 Subtype F1: Where Are We after 33 Years of HIV-1 Infection?Viruses. 2022 Sep 19;14(9):2081. doi: 10.3390/v14092081. Viruses. 2022. PMID: 36146886 Free PMC article. Review.
Cited by
-
Transmission Clusters, Predominantly Associated With Men Who Have Sex With Men, Play a Main Role in the Propagation of HIV-1 in Northern Spain (2013-2018).Front Microbiol. 2022 Mar 31;13:782609. doi: 10.3389/fmicb.2022.782609. eCollection 2022. Front Microbiol. 2022. PMID: 35432279 Free PMC article.
-
Identification of a New HIV-1 BC Intersubtype Circulating Recombinant Form (CRF108_BC) in Spain.Viruses. 2021 Jan 12;13(1):93. doi: 10.3390/v13010093. Viruses. 2021. PMID: 33445523 Free PMC article.
-
Increased HIV Subtype Diversity Reflecting Demographic Changes in the HIV Epidemic in New South Wales, Australia.Viruses. 2020 Dec 6;12(12):1402. doi: 10.3390/v12121402. Viruses. 2020. PMID: 33291330 Free PMC article.
-
Expression of Inflammatory Markers RANK, MMP-9 and PTHrP in Chronic Apical Periodontitis from People Living with HIV Undergoing Antiretroviral Therapy.J Clin Med. 2020 Nov 9;9(11):3611. doi: 10.3390/jcm9113611. J Clin Med. 2020. PMID: 33182451 Free PMC article.
-
Non-B variants of HIV-1 in San Francisco, California.Infect Genet Evol. 2021 Jun;90:104677. doi: 10.1016/j.meegid.2020.104677. Epub 2020 Dec 13. Infect Genet Evol. 2021. PMID: 33321227 Free PMC article.
References
-
- Abecasis A. B., Wensing A. M., Paraskevis D., Vercauteren J., Theys K., van de Vijver D. A., et al. (2013). HIV-1 subtype distribution and its demographic determinants in newly diagnosed patients in Europe suggest highly compartmentalized epidemics. Retrovirology 10:7. 10.1186/1742-4690-10-17 - DOI - PMC - PubMed
LinkOut - more resources
Full Text Sources