

Rare Variants Imputation in Admixed Populations: Comparison Across Reference Panels and Bioinformatics Tools
- PMID: 31001313
- PMCID: PMC6456789
- DOI: 10.3389/fgene.2019.00239
Rare Variants Imputation in Admixed Populations: Comparison Across Reference Panels and Bioinformatics Tools
Abstract
Background: Imputation has become a standard approach in genome-wide association studies (GWAS) to infer in silico untyped markers. Although feasibility for common variants imputation is well established, we aimed to assess rare and ultra-rare variants' imputation in an admixed Caribbean Hispanic population (CH).
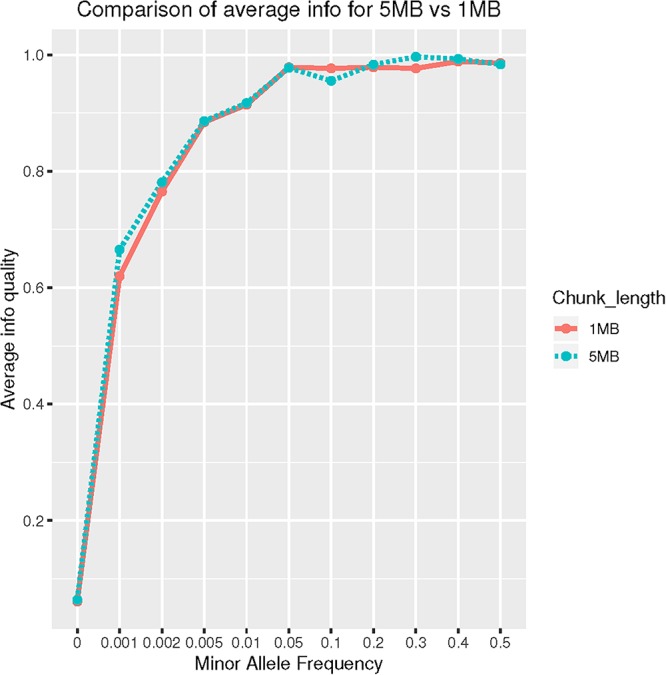
Methods: We evaluated imputation accuracy in CH (N = 1,000), focusing on rare (0.1% ≤ minor allele frequency (MAF) ≤ 1%) and ultra-rare (MAF < 0.1%) variants. We used two reference panels, the Haplotype Reference Consortium (HRC; N = 27,165) and 1000 Genome Project (1000G phase 3; N = 2,504) and multiple phasing (SHAPEIT, Eagle2) and imputation algorithms (IMPUTE2, MACH-Admix). To assess imputation quality, we reported: (a) high-quality variant counts according to imputation tools' internal indexes (e.g., IMPUTE2 "Info" ≥ 80%). (b) Wilcoxon Signed-Rank Test comparing imputation quality for genotyped variants that were masked and imputed; (c) Cohen's kappa coefficient to test agreement between imputed and whole-exome sequencing (WES) variants; (d) imputation of G206A mutation in the PSEN1 (ultra-rare in the general population an more frequent in CH) followed by confirmation genotyping. We also tested ancestry proportion (European, African and Native American) against WES-imputation mismatches in a Poisson regression fashion.
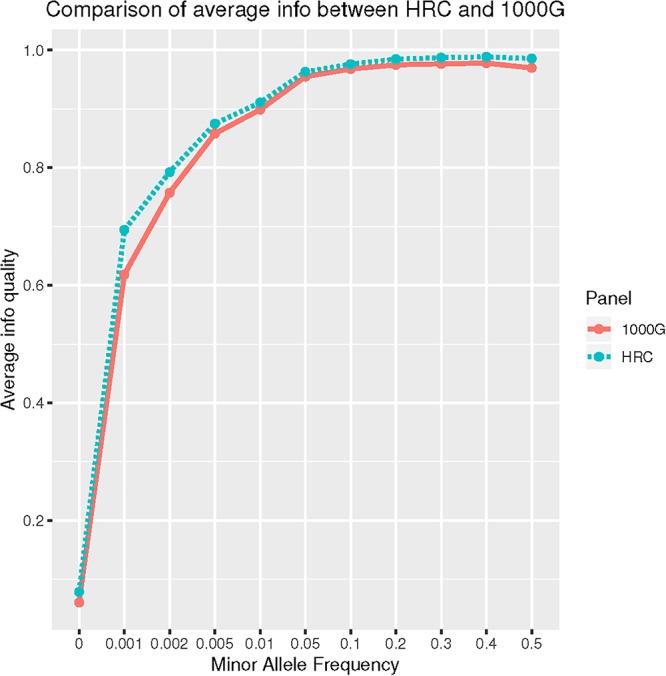
Results: SHAPEIT2 retrieved higher percentage of imputed high-quality variants than Eagle2 (rare: 51.02% vs. 48.60%; ultra-rare 0.66% vs. 0.65%, Wilcoxon p-value < 0.001). SHAPEIT-IMPUTE2 employing HRC outperformed 1000G (64.50% vs. 59.17%; 1.69% vs. 0.75% for high-quality rare and ultra-rare variants, respectively, Wilcoxon p-value < 0.001). SHAPEIT-IMPUTE2 outperformed MaCH-Admix. Compared to 1000G, HRC-imputation retrieved a higher number of high-quality rare and ultra-rare variants, despite showing lower agreement between imputed and WES variants (e.g., rare: 98.86% for HRC vs. 99.02% for 1000G). High Kappa (K = 0.99) was observed for both reference panels. Twelve G206A mutation carriers were imputed and all validated by confirmation genotyping. African ancestry was associated with higher imputation errors for uncommon and rare variants (p-value < 1e-05).
Conclusion: Reference panels with larger numbers of haplotypes can improve imputation quality for rare and ultra-rare variants in admixed populations such as CH. Ethnic composition is an important predictor of imputation accuracy, with higher African ancestry associated with poorer imputation accuracy.
Keywords: 1000G; GWAS; admixed population; imputation; rare variants.
Figures
Similar articles
-
Genotype imputation performance of three reference panels using African ancestry individuals.Hum Genet. 2018 Apr;137(4):281-292. doi: 10.1007/s00439-018-1881-4. Epub 2018 Apr 10. Hum Genet. 2018. PMID: 29637265 Free PMC article.
-
Assessment of genotype imputation performance using 1000 Genomes in African American studies.PLoS One. 2012;7(11):e50610. doi: 10.1371/journal.pone.0050610. Epub 2012 Nov 30. PLoS One. 2012. PMID: 23226329 Free PMC article.
-
Use of >100,000 NHLBI Trans-Omics for Precision Medicine (TOPMed) Consortium whole genome sequences improves imputation quality and detection of rare variant associations in admixed African and Hispanic/Latino populations.PLoS Genet. 2019 Dec 23;15(12):e1008500. doi: 10.1371/journal.pgen.1008500. eCollection 2019 Dec. PLoS Genet. 2019. PMID: 31869403 Free PMC article.
-
Accurate Imputation of Untyped Variants from Deep Sequencing Data.Methods Mol Biol. 2021;2243:271-281. doi: 10.1007/978-1-0716-1103-6_13. Methods Mol Biol. 2021. PMID: 33606262 Review.
-
Evaluation of measures of correctness of genotype imputation in the context of genomic prediction: a review of livestock applications.Animal. 2014 Nov;8(11):1743-53. doi: 10.1017/S1751731114001803. Epub 2014 Jul 21. Animal. 2014. PMID: 25045914 Review.
Cited by
-
Large scale sequence-based screen for recessive variants allows for identification and monitoring of rare deleterious variants in pigs.PLoS Genet. 2024 Jan 10;20(1):e1011034. doi: 10.1371/journal.pgen.1011034. eCollection 2024 Jan. PLoS Genet. 2024. PMID: 38198533 Free PMC article.
-
Polygenic Risk Score for Alzheimer's Disease in Caribbean Hispanics.Ann Neurol. 2021 Sep;90(3):366-376. doi: 10.1002/ana.26131. Epub 2021 Jun 17. Ann Neurol. 2021. PMID: 34038570 Free PMC article.
-
Admixture Mapping of Alzheimer's disease in Caribbean Hispanics identifies a new locus on 22q13.1.Mol Psychiatry. 2022 Jun;27(6):2813-2820. doi: 10.1038/s41380-022-01526-6. Epub 2022 Apr 1. Mol Psychiatry. 2022. PMID: 35365809 Free PMC article.
-
Exploring the role of underrepresented populations in polygenic risk scores for neurodegenerative disease risk prediction.Front Neurosci. 2024 May 27;18:1380860. doi: 10.3389/fnins.2024.1380860. eCollection 2024. Front Neurosci. 2024. PMID: 38859922 Free PMC article. No abstract available.
-
Common genetic risk variants identified in the SPARK cohort support DDHD2 as a candidate risk gene for autism.Transl Psychiatry. 2020 Aug 3;10(1):265. doi: 10.1038/s41398-020-00953-9. Transl Psychiatry. 2020. PMID: 32747698 Free PMC article.
References
Grants and funding
LinkOut - more resources
Full Text Sources