

Radiotherapy Both Promotes and Inhibits Myeloid-Derived Suppressor Cell Function: Novel Strategies for Preventing the Tumor-Protective Effects of Radiotherapy
- PMID: 31001479
- PMCID: PMC6454107
- DOI: 10.3389/fonc.2019.00215
Radiotherapy Both Promotes and Inhibits Myeloid-Derived Suppressor Cell Function: Novel Strategies for Preventing the Tumor-Protective Effects of Radiotherapy
Abstract
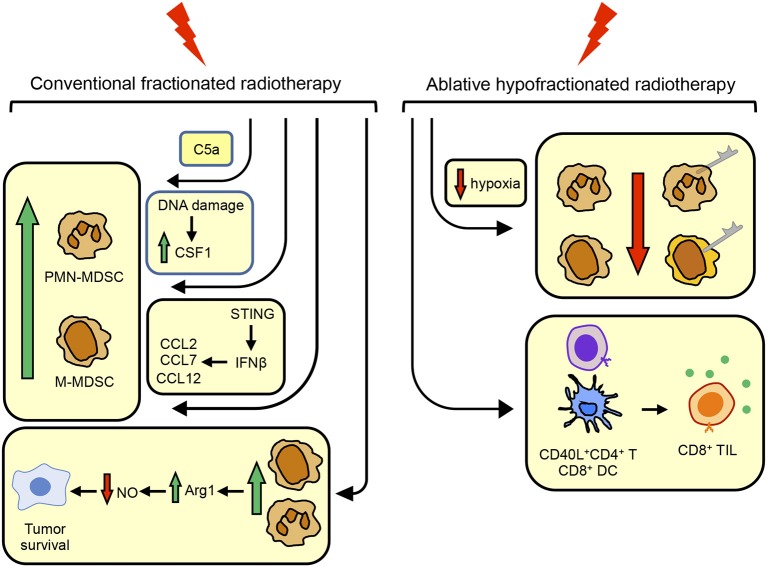
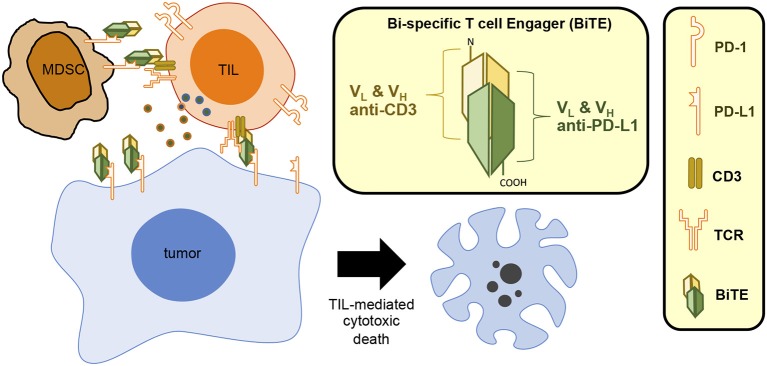
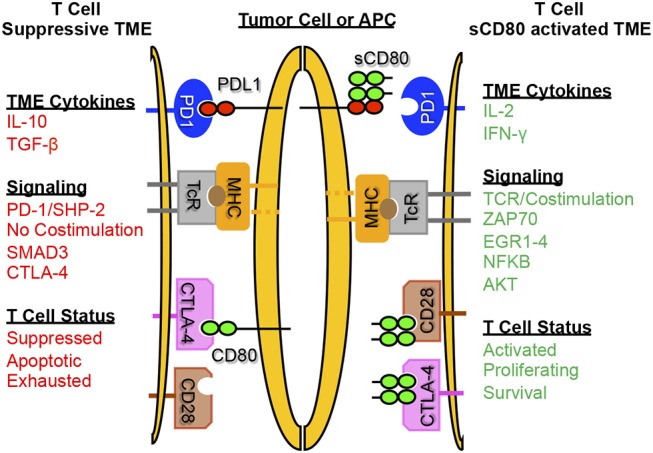
Cancer immunotherapies aimed at neutralizing the programmed death-1 (PD-1) immune suppressive pathway have yielded significant therapeutic efficacy in a subset of cancer patients. However, only a subset of patients responds to antibody therapy with either anti-PD-1 or anti-PD-L1 antibodies. These patients appear to have so-called "hot" tumors containing tumor-reactive T cells. Therefore, checkpoint blockade therapy may be effective in a larger percentage of cancer patients if combined with therapeutics that also activate tumor-reactive T cells. Radiotherapy (RT) is a prime candidate for combination therapy because it facilitates activation of both local antitumor immunity and antitumor immunity at non-radiated, distant sites (abscopal response). However, RT also promotes tumor cell expression of PD-L1 and facilitates the development of myeloid-derived suppressor cells (MDSC), a population of immune suppressive cells that also suppress through PD-L1. This article will review how RT induces MDSC, and then describe two novel therapeutics that are designed to simultaneously activate tumor-reactive T cells and neutralize PD-1-mediated immune suppression. One therapeutic, a CD3xPD-L1 bispecific T cell engager (BiTE), activates and targets cytotoxic T and NKT cells to kill PD-L1+ tumor cells, despite the presence of MDSC. The BiTE significantly extends the survival time of humanized NSG mice reconstituted with human PBMC and carrying established metastatic human melanoma tumors. The second therapeutic is a soluble form of the costimulatory molecule CD80 (sCD80). In addition to costimulating through CD28, sCD80 inhibits PD-1 suppression by binding to PD-L1 and sterically blocking PD-L1/PD-1 signaling. sCD80 increases tumor-infiltrating T cells and significantly extends survival time of mice carrying established, syngeneic tumors. sCD80 does not suppress T cell function via CTLA-4. These studies suggest that the CD3xPD-L1 BiTE and sCD80 may be efficacious therapeutics either as monotherapies or in combination with other therapies such as radiation therapy for the treatment of cancer.
Keywords: bi-specific T cell engager (BiTE); myeloid-derived suppressor cells (MDSC); programmed death ligand 1 (PD-L1); radiotherapy-induced immune suppression; solubilized CD80.
Figures
References
Publication types
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials