

Genetic landscape and macro-evolution of co-circulating Coxsackieviruses A and Vaccine-derived Polioviruses in the Democratic Republic of Congo, 2008-2013
- PMID: 31002713
- PMCID: PMC6505894
- DOI: 10.1371/journal.pntd.0007335
Genetic landscape and macro-evolution of co-circulating Coxsackieviruses A and Vaccine-derived Polioviruses in the Democratic Republic of Congo, 2008-2013
Abstract
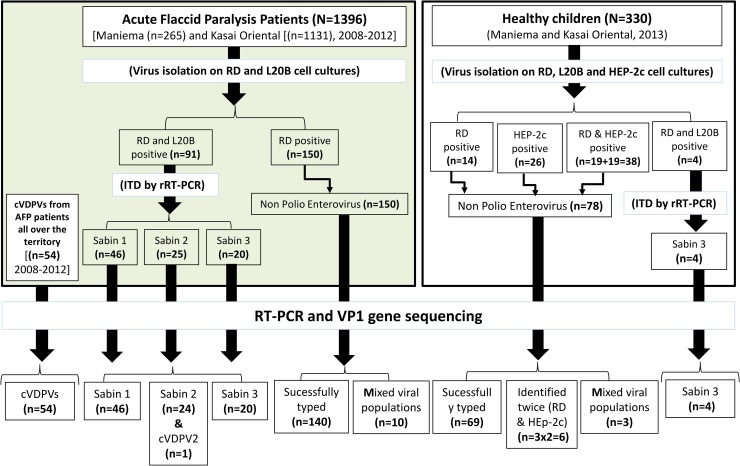
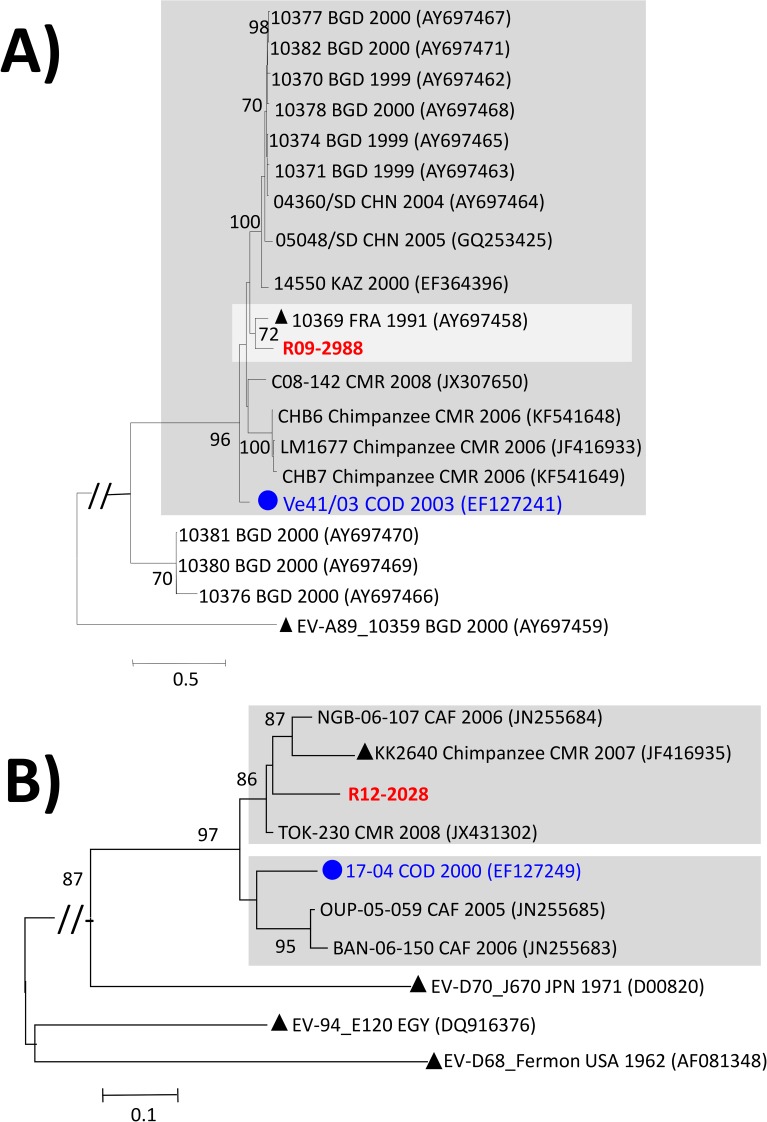
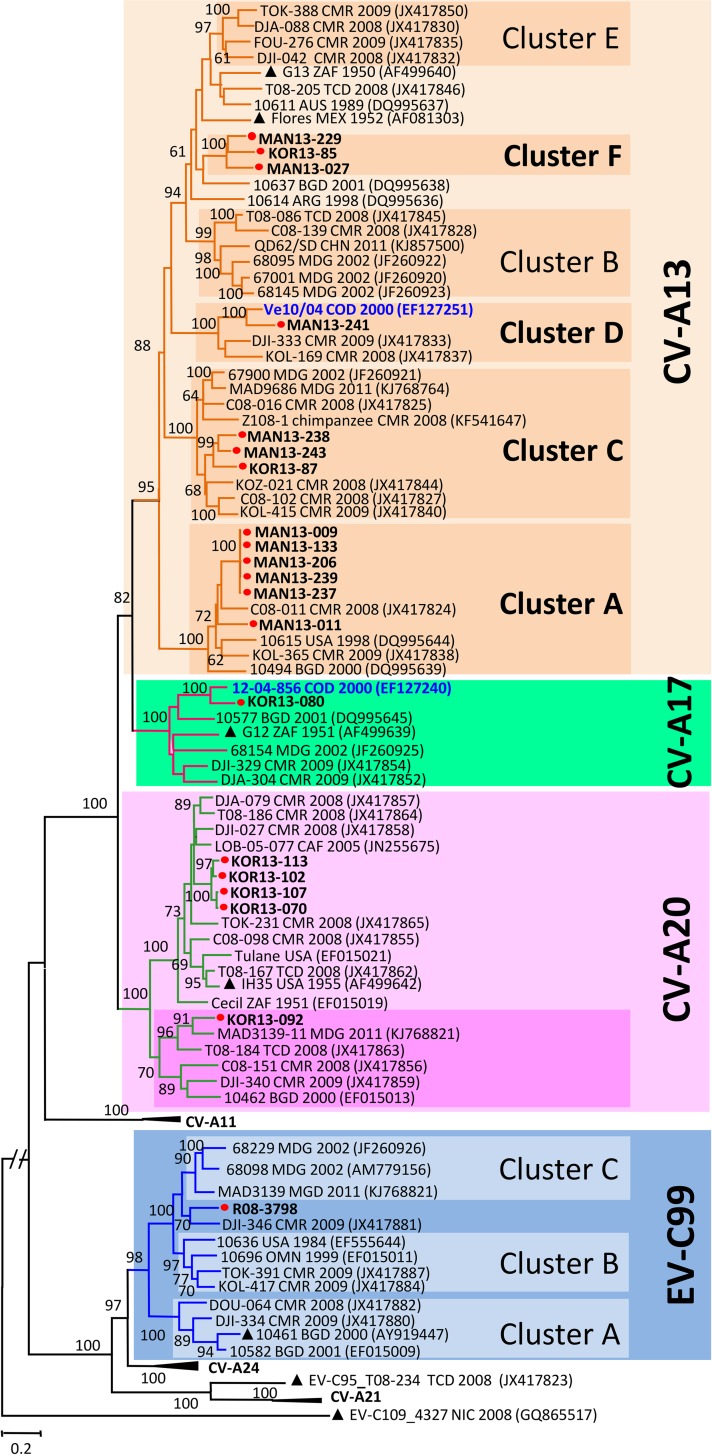
Enteroviruses (EVs) are among the most common viruses infecting humans worldwide but only a few Non-Polio Enterovirus (NPEV) isolates have been characterized in the Democratic Republic of Congo (DR Congo). Moreover, circulating vaccine-derived polioviruses (PVs) [cVDPVs] isolated during multiple outbreaks in DR Congo from 2004 to 2018 have been characterized so far only by the sequences of their VP1 capsid coding gene. This study was carried to i) investigate the circulation and genetic diversity of NPEV and polio vaccine isolates recovered from healthy children and Acute Flaccid Paralysis (AFP) patients, ii) evaluate the occurrence of genetic recombination among EVs belonging to the Enterovirus C species (including PVs) and iii) identify the virological factors favoring multiple emergences of cVDPVs in DR Congo. The biological material considered in this study included i) a collection of 91 Sabin-like PVs, 54 cVDPVs and 150 NPEVs isolated from AFP patients between 2008 and 2012 in DR Congo and iii) a collection of 330 stool specimens collected from healthy children in 2013 in the Kasai Oriental and Maniema provinces of DR Congo. Studied virus isolates were sequenced in four distinct sub-genomic regions 5'-UTR, VP1, 2CATPase and 3Dpol. Resulting sequences were compared through comparative phylogenetic analyses. Virus isolation showed that 19.1% (63/330) healthy children were infected by EVs including 17.9% (59/330) of NPEVs and 1.2% (4/330) of type 3 Sabin-like PVs. Only one EV-C type, EV-C99 was identified among the NPEV collection from AFP patients whereas 27.5% of the 69 NPEV isolates typed in healthy children belonged to the EV-C species: CV-A13 (13/69), A20 (5/69) and A17 (1/69). Interestingly, 50 of the 54 cVDPVs featured recombinant genomes containing exogenous sequences in at least one of the targeted non-structural regions of their genomes: 5'UTR, 2CATPase and 3Dpol. Some of these non-vaccine sequences of the recombinant cVDPVs were strikingly related to homologous sequences from co-circulating CV-A17 and A20 in the 2CATPase region as well as to those from co-circulating CV-A13, A17 and A20 in the 3Dpol region. This study provided the first evidence uncovering CV-A20 strains as major recombination partners of PVs. High quality AFP surveillance, sensitive environmental surveillance and efficient vaccination activities remain essential to ensure timely detection and efficient response to recombinant cVDPVs outbreaks in DR Congo. Such needs are valid for any epidemiological setting where high frequency and genetic diversity of Coxsackieviruses A13, A17 and A20 provide a conducive viral ecosystem for the emergence of virulent recombinant cVDPVs.
Conflict of interest statement
The authors have declared that no competing interests exist.
Figures


References
-
- Marx A, Glass JD, Sutter RW. Differential diagnosis of acute flaccid paralysis and its role in poliomyelitis surveillance. Epidemiol Rev. 2000;22(2):298–316. - PubMed
-
- Willison HJ, Jacobs BC, van Doorn PA. Guillain-Barre syndrome. Lancet. 2016;29(16):00339–1. - PubMed
-
- Knowles N. Picornaviridae Study Group website. Available at: http://www.picornaviridae.com/enterovirus/enterovirus.htm (accessed February 25, 2019).
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Research Materials
