

Protein Sequence Editing of SKN-1A/Nrf1 by Peptide:N-Glycanase Controls Proteasome Gene Expression
- PMID: 31002798
- PMCID: PMC6574124
- DOI: 10.1016/j.cell.2019.03.035
Protein Sequence Editing of SKN-1A/Nrf1 by Peptide:N-Glycanase Controls Proteasome Gene Expression
Abstract
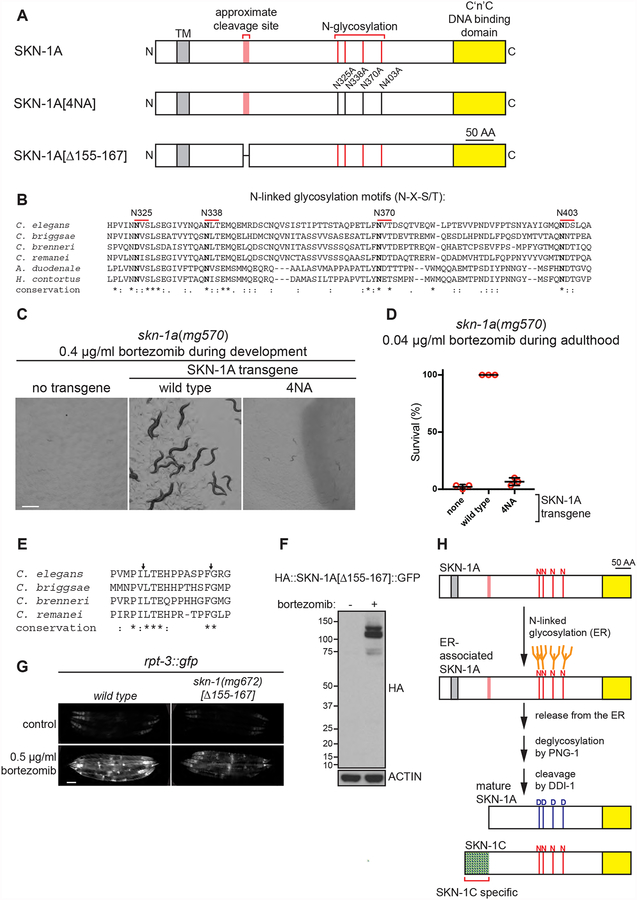
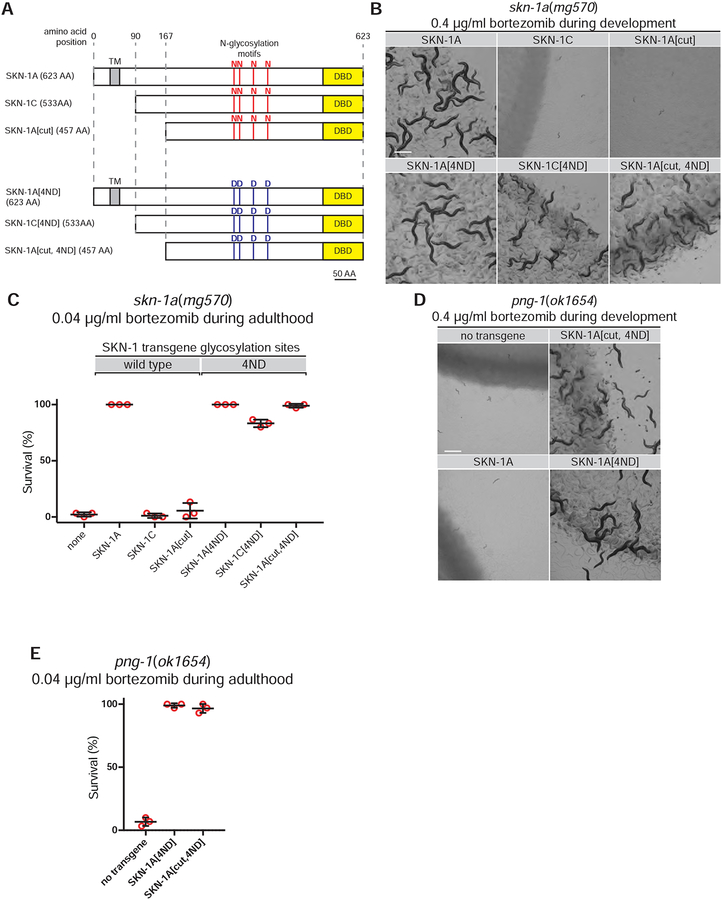
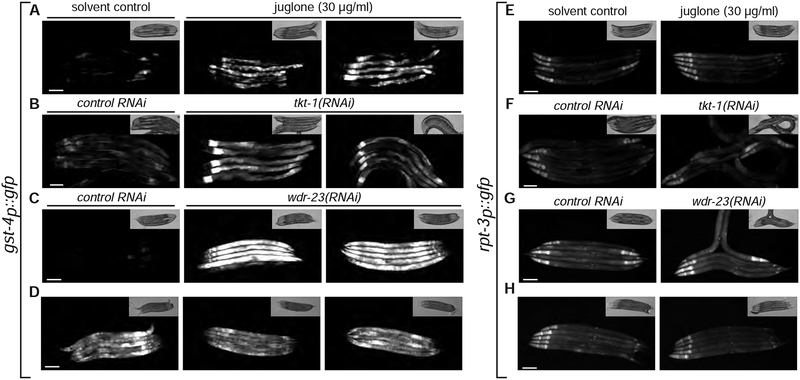
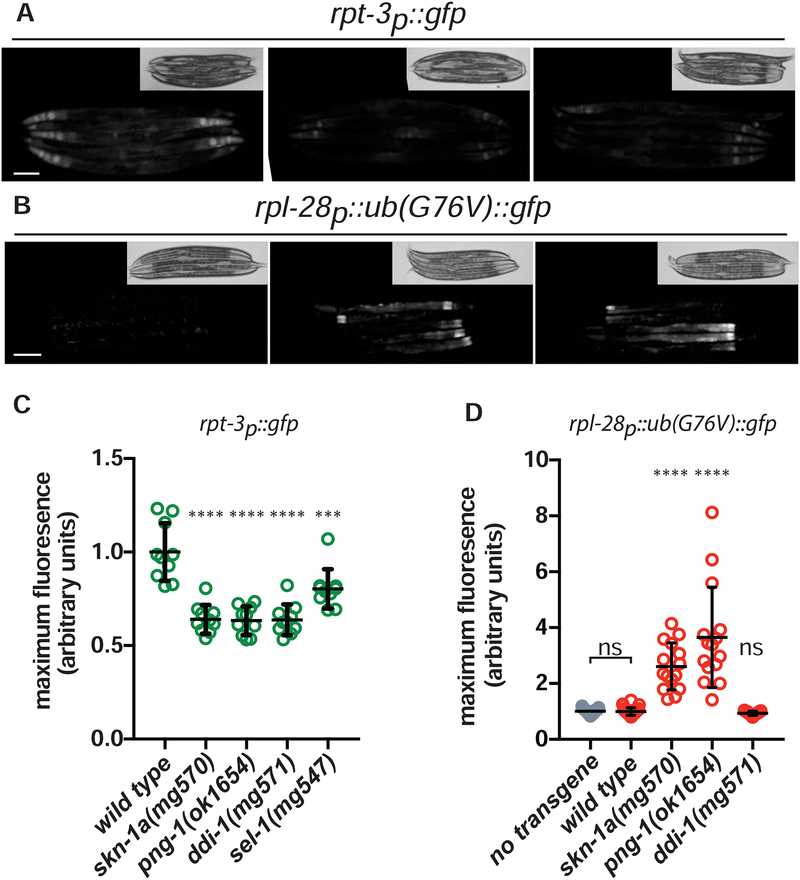
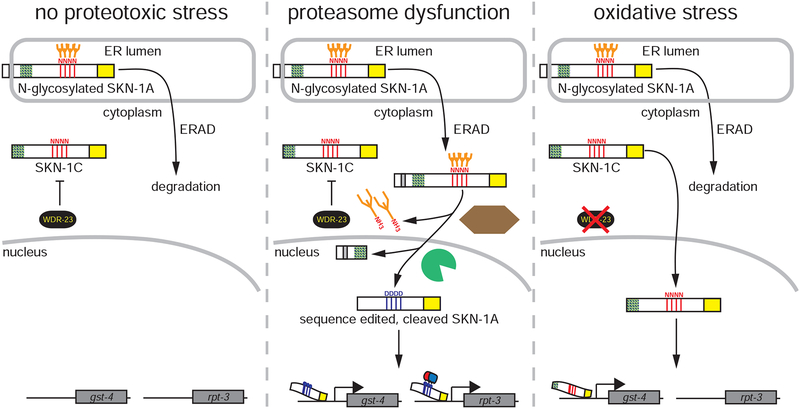
The proteasome mediates selective protein degradation and is dynamically regulated in response to proteotoxic challenges. SKN-1A/Nrf1, an endoplasmic reticulum (ER)-associated transcription factor that undergoes N-linked glycosylation, serves as a sensor of proteasome dysfunction and triggers compensatory upregulation of proteasome subunit genes. Here, we show that the PNG-1/NGLY1 peptide:N-glycanase edits the sequence of SKN-1A protein by converting particular N-glycosylated asparagine residues to aspartic acid. Genetically introducing aspartates at these N-glycosylation sites bypasses the requirement for PNG-1/NGLY1, showing that protein sequence editing rather than deglycosylation is key to SKN-1A function. This pathway is required to maintain sufficient proteasome expression and activity, and SKN-1A hyperactivation confers resistance to the proteotoxicity of human amyloid beta peptide. Deglycosylation-dependent protein sequence editing explains how ER-associated and cytosolic isoforms of SKN-1 perform distinct cytoprotective functions corresponding to those of mammalian Nrf1 and Nrf2. Thus, we uncover an unexpected mechanism by which N-linked glycosylation regulates protein function and proteostasis.
Keywords: N-linked glycosylation; NFE2L1; NFE2L2; NGLY1; NGLY1 deficiency; Nrf1; Nrf2; PNG-1; PNGase; SKN-1; SKN-1A; bortezomib; deglycosylation; glycobiology; neurodegenerative diseases; peptide:N-glycanase; proteasome; protein quality control; protein sequence editing; proteostasis.
Copyright © 2019 Elsevier Inc. All rights reserved.
Conflict of interest statement
DECLARATION OF INTERESTS
The authors have no competing interests to declare.
Figures

Comment in
-
Editing proteasome synthesis.Nat Rev Mol Cell Biol. 2019 Jun;20(6):324-325. doi: 10.1038/s41580-019-0140-4. Nat Rev Mol Cell Biol. 2019. PMID: 31053827 No abstract available.
References
-
- Bishop NA, and Guarente L (2007). Two neurons mediate diet-restriction-induced longevity in C. elegans. Nature 447, 545–549. - PubMed
-
- Blackwell TK, Bowerman B, Priess JR, and Weintraub H (1994). Formation of a monomeric DNA binding domain by Skn-1 bZIP and homeodomain elements. Science 266, 621–628. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
