

Delta-secretase-cleaved Tau antagonizes TrkB neurotrophic signalings, mediating Alzheimer's disease pathologies
- PMID: 31004063
- PMCID: PMC6500177
- DOI: 10.1073/pnas.1901348116
Delta-secretase-cleaved Tau antagonizes TrkB neurotrophic signalings, mediating Alzheimer's disease pathologies
Abstract
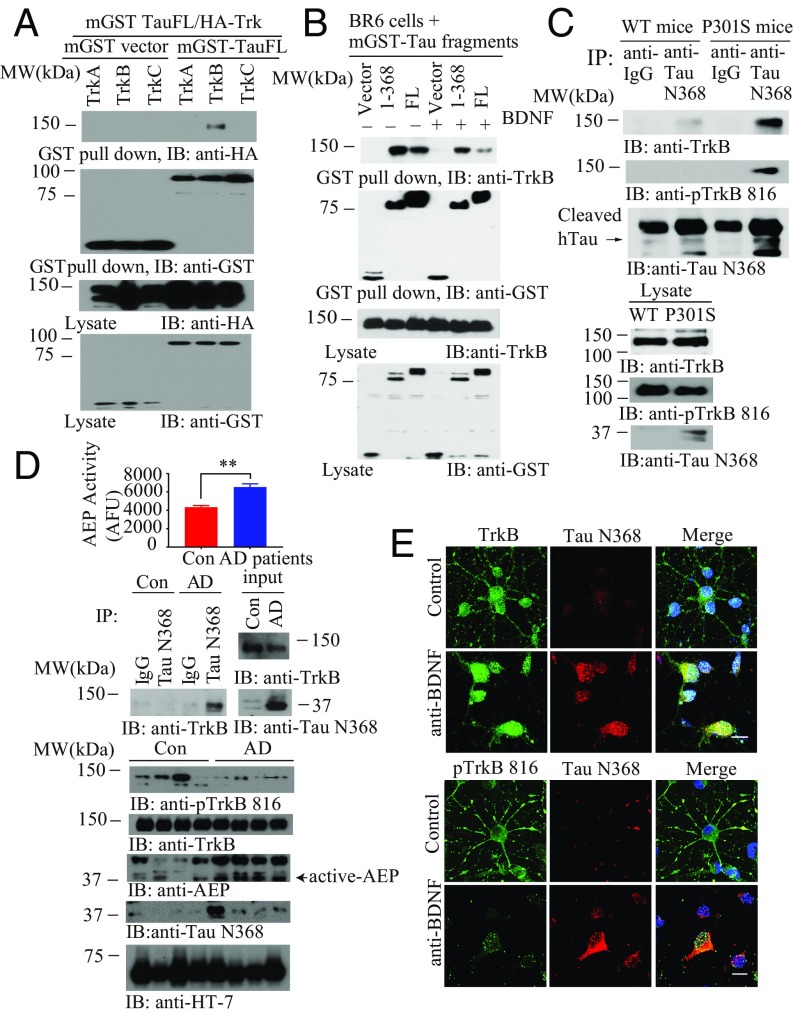
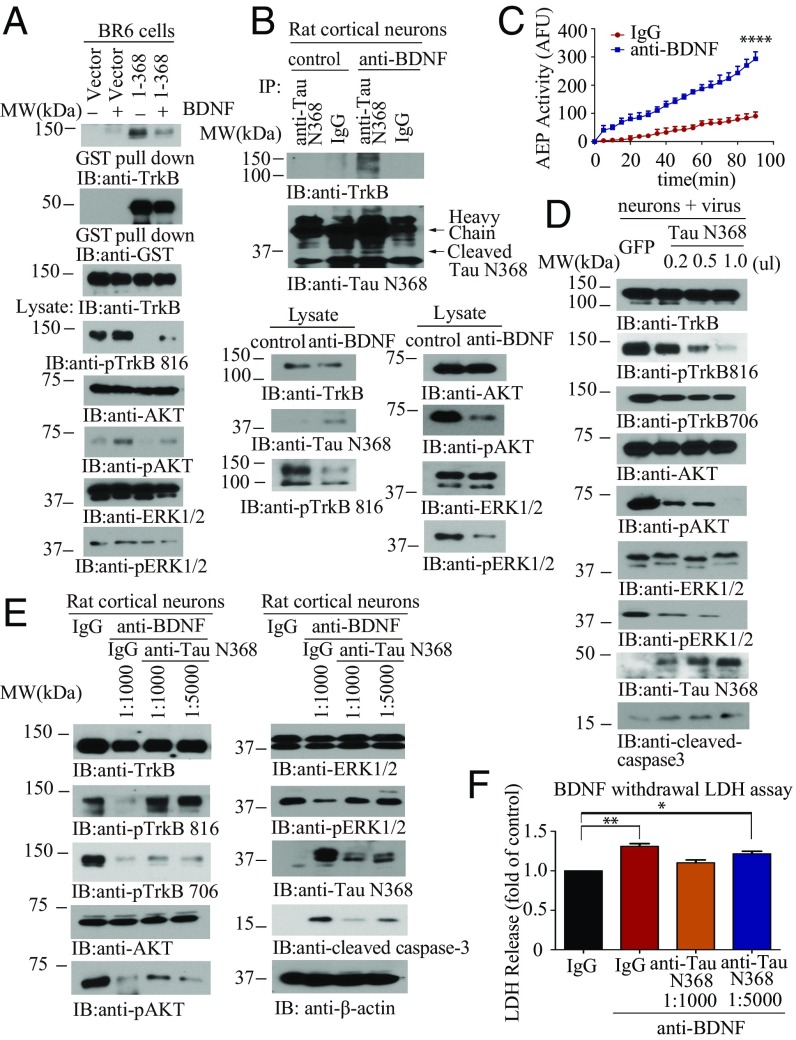
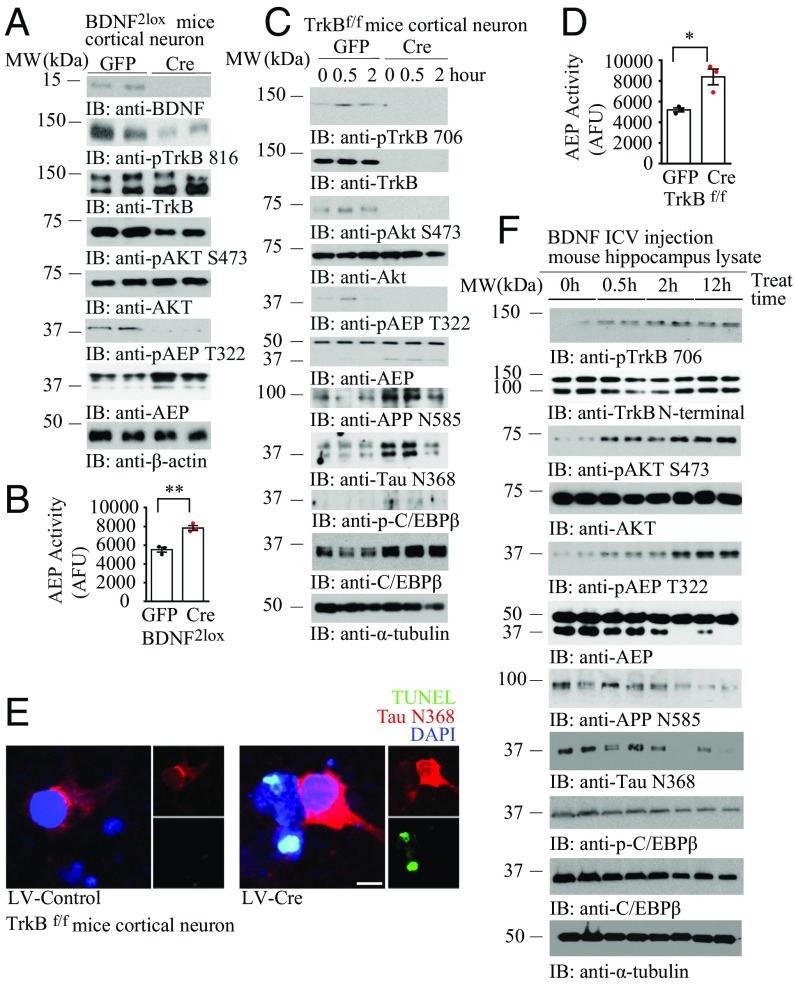
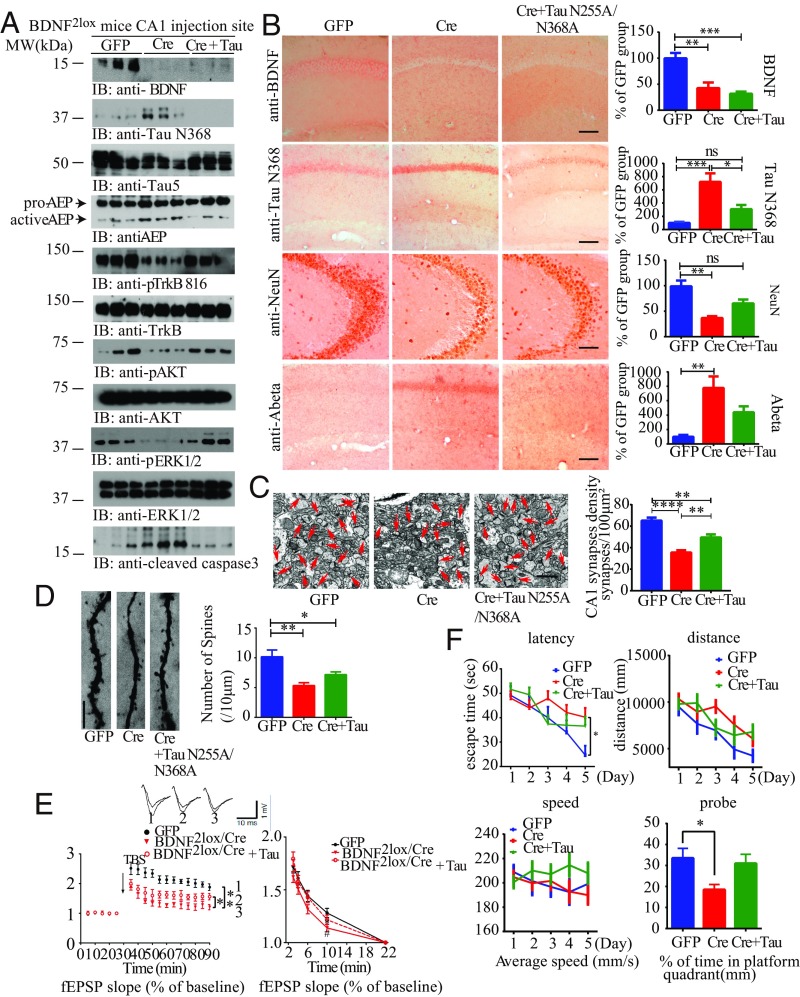
BDNF, an essential trophic factor implicated in synaptic plasticity and neuronal survival, is reduced in Alzheimer's disease (AD). BDNF deficiency's association with Tau pathology in AD is well documented. However, the molecular mechanisms accounting for these events remain incompletely understood. Here we show that BDNF deprivation triggers Tau proteolytic cleavage by activating δ-secretase [i.e., asparagine endopeptidase (AEP)], and the resultant Tau N368 fragment binds TrkB receptors and blocks its neurotrophic signals, inducing neuronal cell death. Knockout of BDNF or TrkB receptors provokes δ-secretase activation via reducing T322 phosphorylation by Akt and subsequent Tau N368 cleavage, inducing AD-like pathology and cognitive dysfunction, which can be restored by expression of uncleavable Tau N255A/N368A mutant. Blocking the Tau N368-TrkB complex using Tau repeat-domain 1 peptide reverses this pathology. Thus, our findings support that BDNF reduction mediates Tau pathology via activating δ-secretase in AD.
Keywords: AEP; Alzheimer’s disease; BDNF deprivation; Tau N368; Tauopathy.
Conflict of interest statement
The authors declare no conflict of interest.
Figures

References
-
- Minichiello L. TrkB signalling pathways in LTP and learning. Nat Rev Neurosci. 2009;10:850–860. - PubMed
-
- Ferrer I, et al. BDNF and full-length and truncated TrkB expression in Alzheimer disease. Implications in therapeutic strategies. J Neuropathol Exp Neurol. 1999;58:729–739. - PubMed
-
- Garzon D, Yu G, Fahnestock M. A new brain-derived neurotrophic factor transcript and decrease in brain-derived neurotrophic factor transcripts 1, 2 and 3 in Alzheimer’s disease parietal cortex. J Neurochem. 2002;82:1058–1064. - PubMed
-
- Ginsberg SD, Che S, Wuu J, Counts SE, Mufson EJ. Down regulation of trk but not p75NTR gene expression in single cholinergic basal forebrain neurons mark the progression of Alzheimer’s disease. J Neurochem. 2006;97:475–487. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
