

Redox control of non-shivering thermogenesis
- PMID: 31005563
- PMCID: PMC6599457
- DOI: 10.1016/j.molmet.2019.04.002
Redox control of non-shivering thermogenesis
Abstract
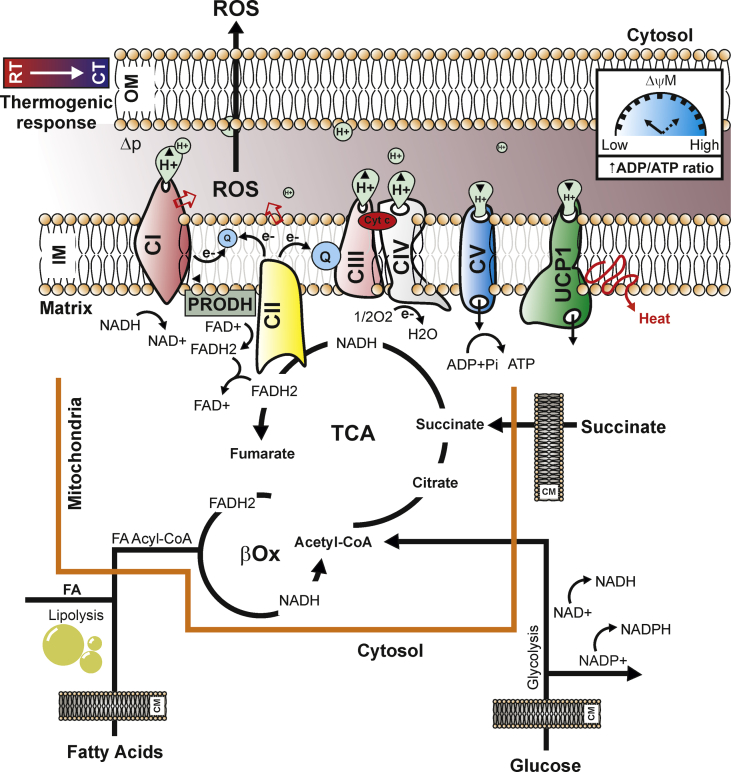
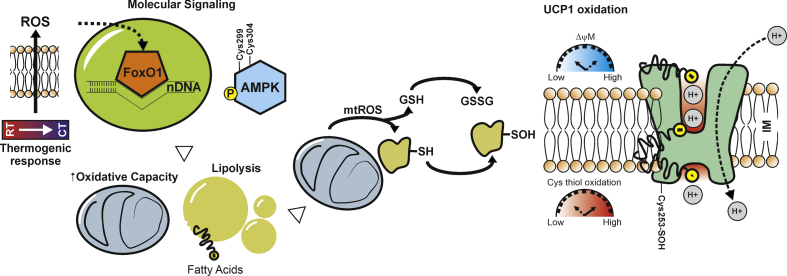
Background: Thermogenic adipocytes reorganize their metabolism during cold exposure. Metabolic reprogramming requires readily available bioenergetics substrates, such as glucose and fatty acids, to increase mitochondrial respiration and produce heat via the uncoupling protein 1 (UCP1). This condition generates a finely-tuned production of mitochondrial reactive oxygen species (ROS) that support non-shivering thermogenesis.
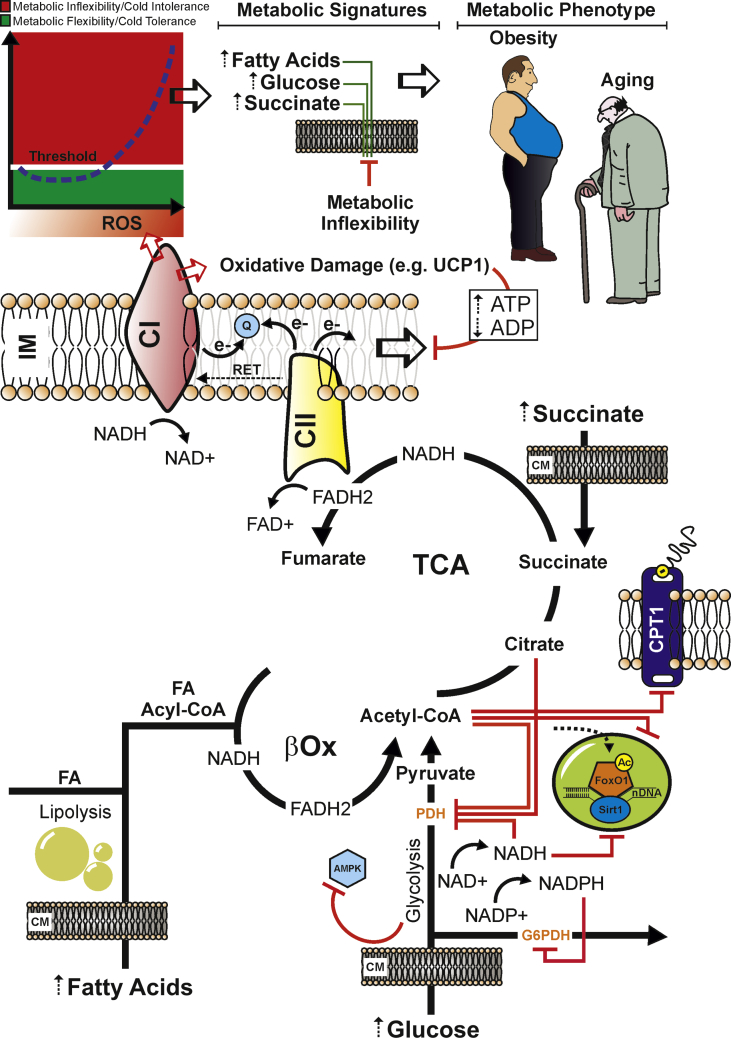
Scope of review: Herein, the findings underlining the mechanisms that regulate ROS production and control of the adaptive responses tuning thermogenesis in adipocytes are described. Furthermore, this review describes the metabolic responses to substrate availability and the consequence of mitochondrial failure to switch fuel oxidation in response to changes in nutrient availability. A framework to control mitochondrial ROS threshold to maximize non-shivering thermogenesis in adipocytes is provided.
Major conclusions: Thermogenesis synchronizes fuel oxidation with an acute and transient increase of mitochondrial ROS that promotes the activation of redox-sensitive thermogenic signaling cascade and UCP1. However, an overload of substrate flux to mitochondria causes a massive and damaging mitochondrial ROS production that affects mitochondrial flexibility. Finding novel thermogenic redox targets and manipulating ROS concentration in adipocytes appears to be a promising avenue of research for improving thermogenesis and counteracting metabolic diseases.
Keywords: Adipocyte; Adipose tissue; Mitochondrial metabolism; Obesity; Type 2 diabetes.
Copyright © 2019 The Author. Published by Elsevier GmbH.. All rights reserved.
Figures
References
-
- Chouchani E.T., Kazak L., Spiegelman B.M. New advances in adaptive thermogenesis: UCP1 and beyond. Cell Metabolism. 2019;29:27–37. - PubMed
-
- Jastroch M., Oelkrug R., Keipert S. Insights into brown adipose tissue evolution and function from non-model organisms. Journal of Experimental Biology. 2018;221 - PubMed
-
- Genin F., Nibbelink M., Galand M., Perret M., Ambid L. Brown fat and nonshivering thermogenesis in the gray mouse lemur (Microcebus murinus) American Journal of Physiology – Regulatory, Integrative and Comparative Physiology. 2003;284:R811–R818. - PubMed
-
- van Marken Lichtenbelt W.D., Vanhommerig J.W., Smulders N.M., Drossaerts J.M., Kemerink G.J., Bouvy N.D. Cold-activated brown adipose tissue in healthy men. New England Journal of Medicine. 2009;360:1500–1508. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Research Materials
