

End-to-End Differentiable Learning of Protein Structure
- PMID: 31005579
- PMCID: PMC6513320
- DOI: 10.1016/j.cels.2019.03.006
End-to-End Differentiable Learning of Protein Structure
Abstract
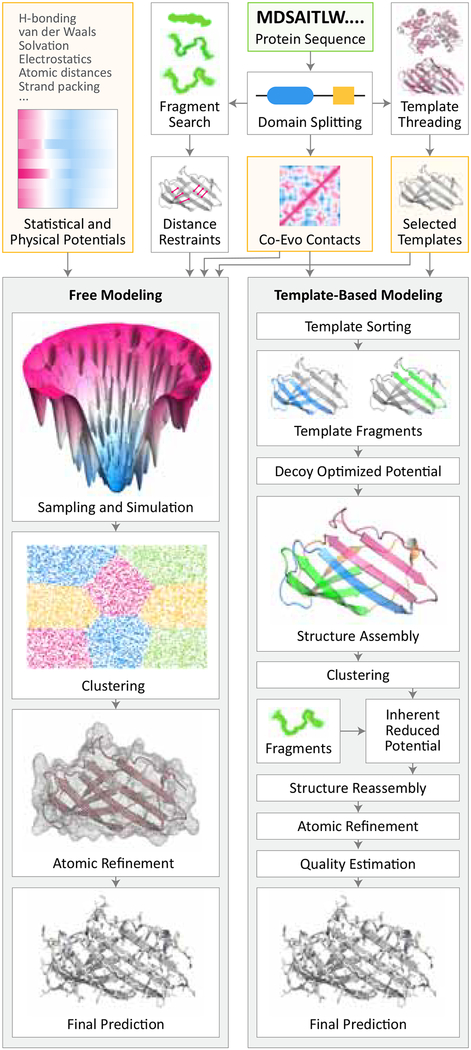
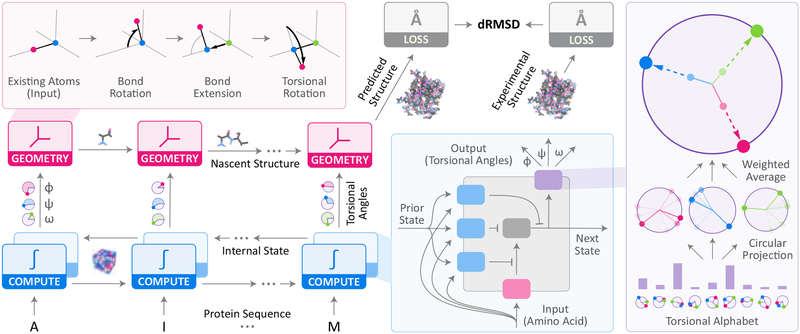
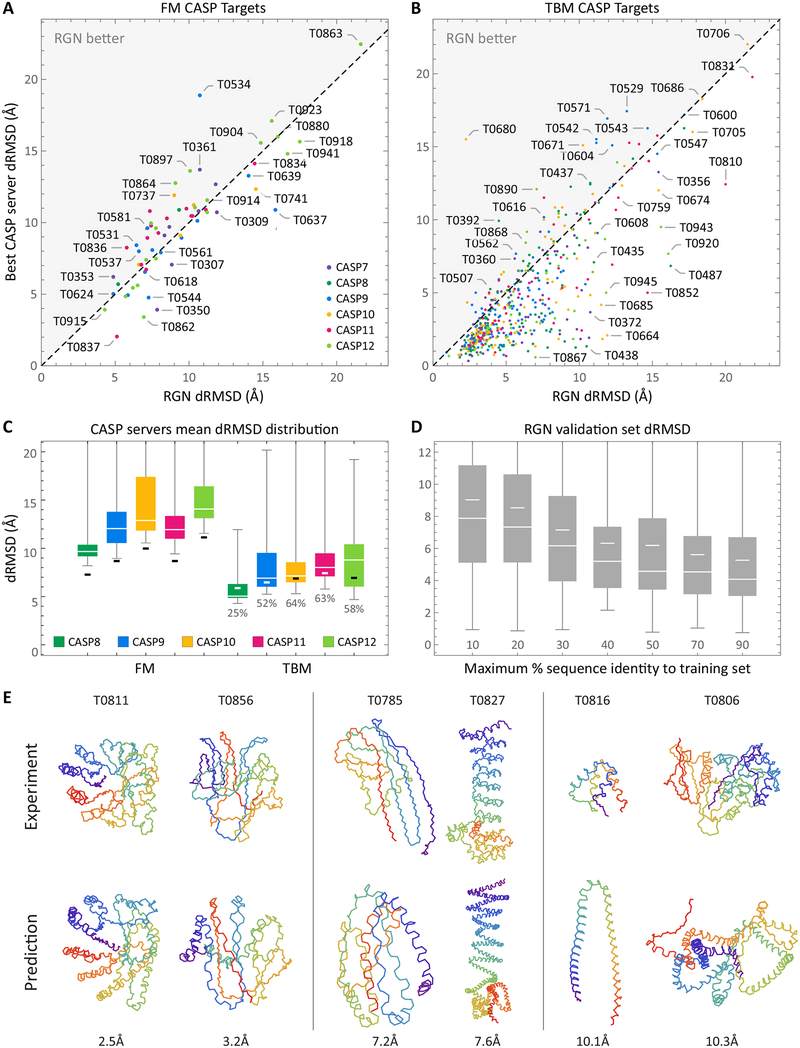
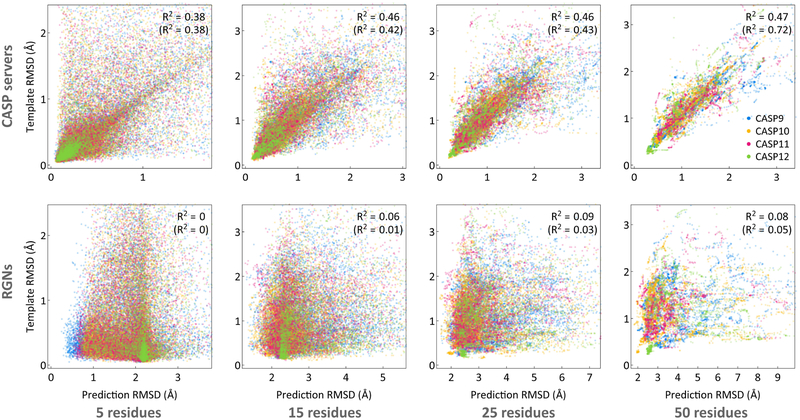
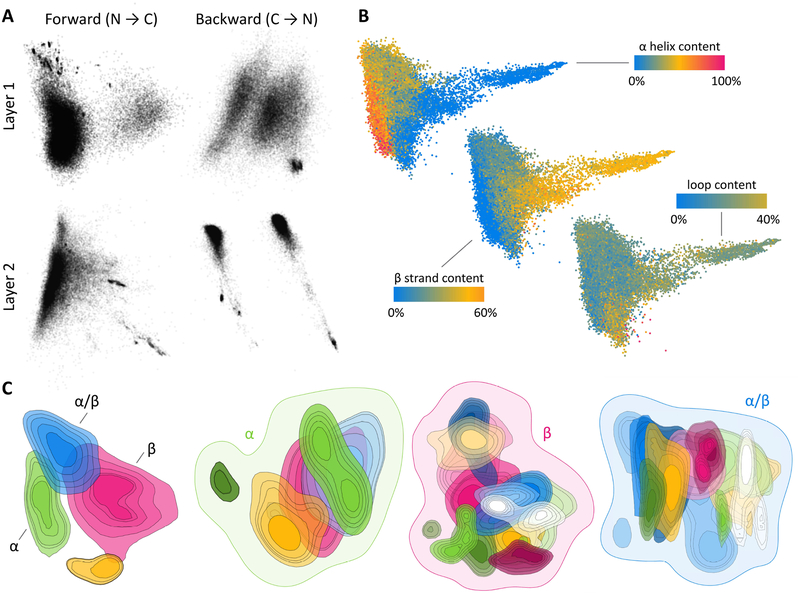
Predicting protein structure from sequence is a central challenge of biochemistry. Co-evolution methods show promise, but an explicit sequence-to-structure map remains elusive. Advances in deep learning that replace complex, human-designed pipelines with differentiable models optimized end to end suggest the potential benefits of similarly reformulating structure prediction. Here, we introduce an end-to-end differentiable model for protein structure learning. The model couples local and global protein structure via geometric units that optimize global geometry without violating local covalent chemistry. We test our model using two challenging tasks: predicting novel folds without co-evolutionary data and predicting known folds without structural templates. In the first task, the model achieves state-of-the-art accuracy, and in the second, it comes within 1-2 Å; competing methods using co-evolution and experimental templates have been refined over many years, and it is likely that the differentiable approach has substantial room for further improvement, with applications ranging from drug discovery to protein design.
Keywords: biophysics; co-evolution; deep learning; geometric deep learning; homology modeling; machine learning; protein design; protein folding; protein structure prediction; structural biology.
Copyright © 2019 Elsevier Inc. All rights reserved.
Conflict of interest statement
Declaration of Interests:
The author declares no competing interests.
Figures
References
-
- Abadi M, Barham P, Chen J, Chen Z, Davis A, Dean J, Devin M, Ghemawat S, Irving G, Isard M, et al. (2016). TensorFlow: A system for large-scale machine learning In 12th USENIX Symposium on Operating Systems Design and Implementation (OSDI 16), pp. 265–283.
-
- Alain G, and Bengio Y (2016). Understanding intermediate layers using linear classifier probes. ArXiv:1610.01644 [Cs, Stat].
-
- AlQuraishi M (2019a). Parallelized Natural Extension Reference Frame: Parallelized Conversion from Internal to Cartesian Coordinates. Journal of Computational Chemistry 40, 885–892. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
