

Preclinical Evaluation of Allogeneic CAR T Cells Targeting BCMA for the Treatment of Multiple Myeloma
- PMID: 31005597
- PMCID: PMC6554542
- DOI: 10.1016/j.ymthe.2019.04.001
Preclinical Evaluation of Allogeneic CAR T Cells Targeting BCMA for the Treatment of Multiple Myeloma
Abstract
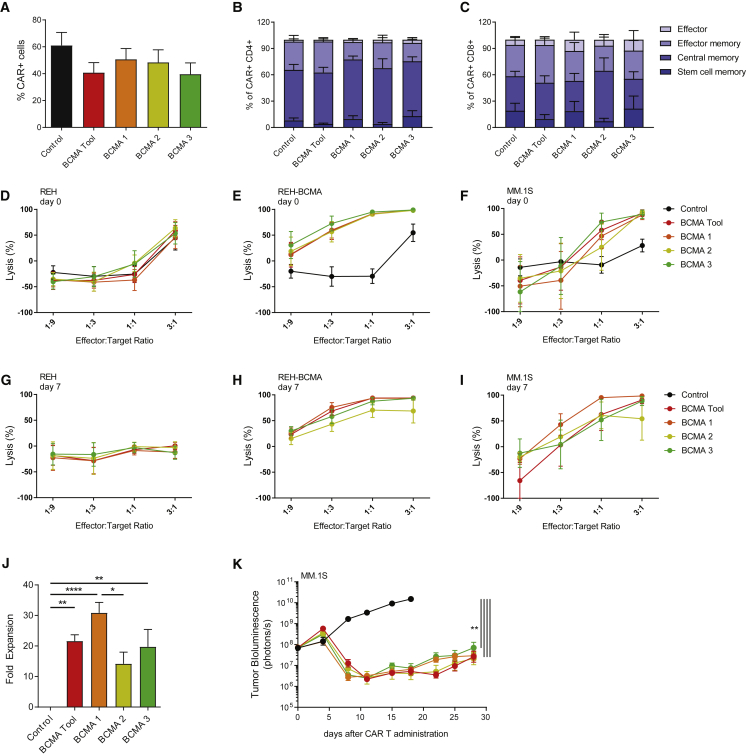
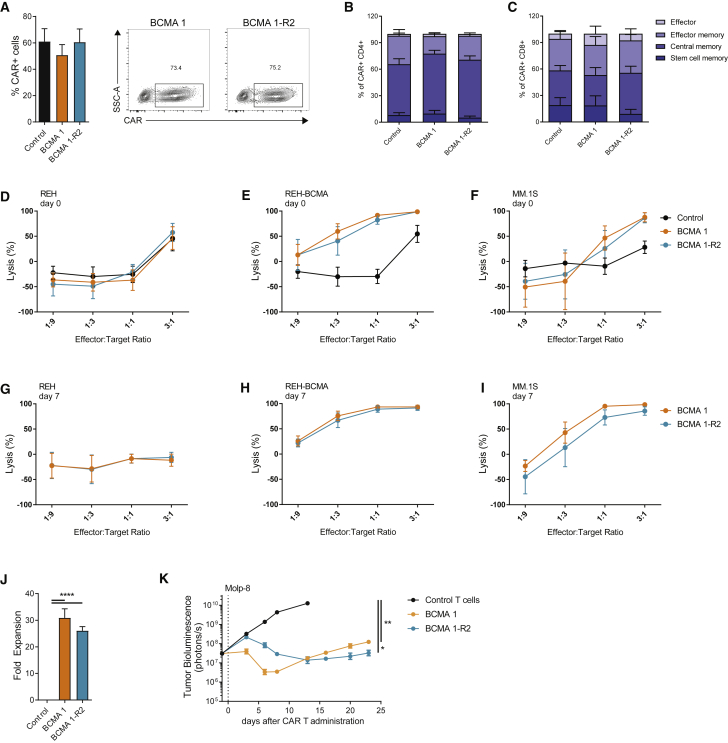
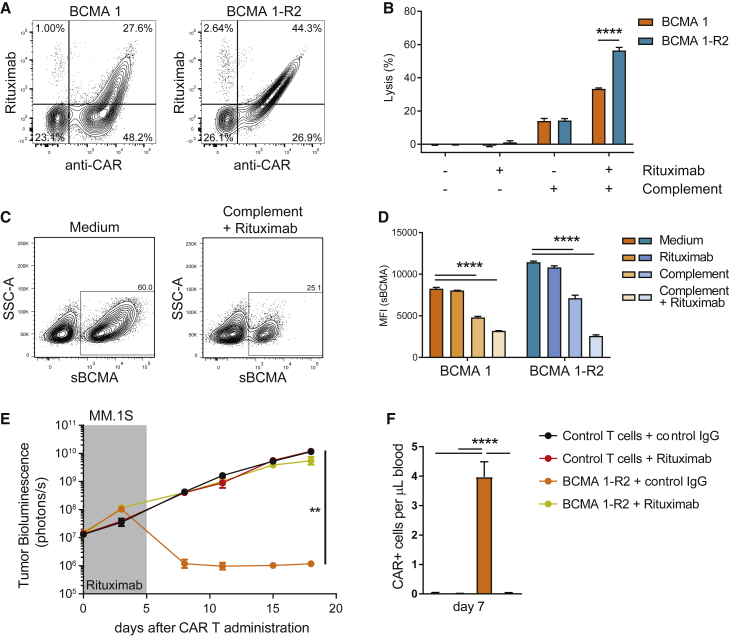
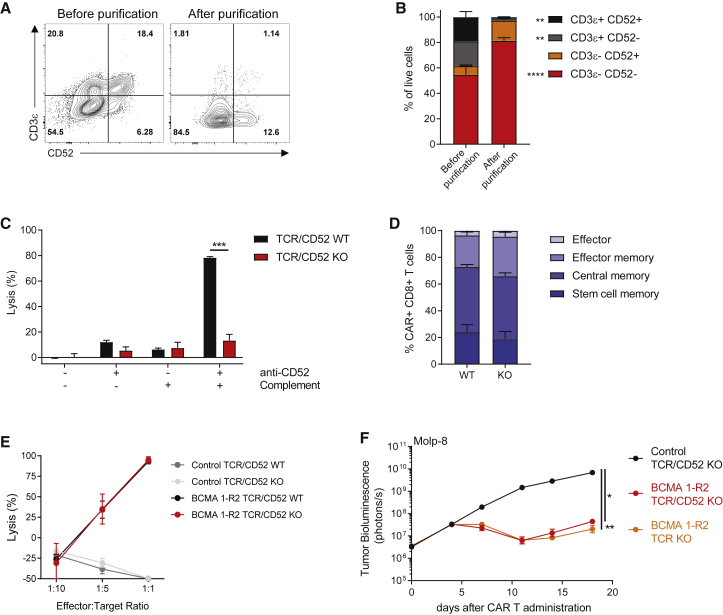
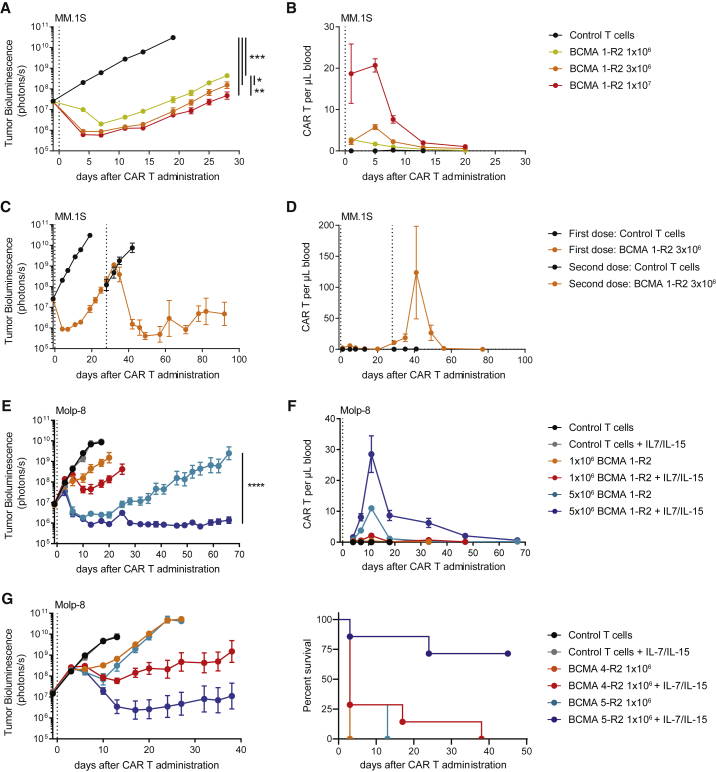
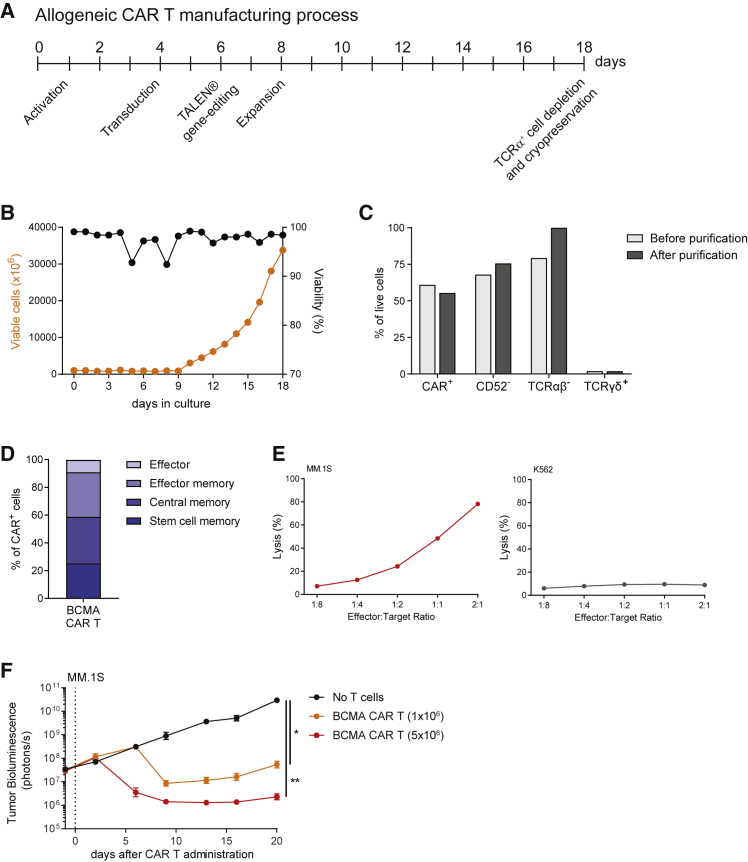
Clinical success of autologous CD19-directed chimeric antigen receptor T cells (CAR Ts) in acute lymphoblastic leukemia and non-Hodgkin lymphoma suggests that CAR Ts may be a promising therapy for hematological malignancies, including multiple myeloma. However, autologous CAR T therapies have limitations that may impact clinical use, including lengthy vein-to-vein time and manufacturing constraints. Allogeneic CAR T (AlloCAR T) therapies may overcome these innate limitations of autologous CAR T therapies. Unlike autologous cell therapies, AlloCAR T therapies employ healthy donor T cells that are isolated in a manufacturing facility, engineered to express CARs with specificity for a tumor-associated antigen, and modified using gene-editing technology to limit T cell receptor (TCR)-mediated immune responses. Here, transcription activator-like effector nuclease (TALEN) gene editing of B cell maturation antigen (BCMA) CAR Ts was used to confer lymphodepletion resistance and reduced graft-versus-host disease (GvHD) potential. The safety profile of allogeneic BCMA CAR Ts was further enhanced by incorporating a CD20 mimotope-based intra-CAR off switch enabling effective CAR T elimination in the presence of rituximab. Allogeneic BCMA CAR Ts induced sustained antitumor responses in mice supplemented with human cytokines, and, most importantly, maintained their phenotype and potency after scale-up manufacturing. This novel off-the-shelf allogeneic BCMA CAR T product is a promising candidate for clinical evaluation.
Keywords: AlloCAR T; B cell maturation antigen; allogeneic CAR T therapy; chimeric antigen receptor; multiple myeloma.
Copyright © 2019 The American Society of Gene and Cell Therapy. Published by Elsevier Inc. All rights reserved.
Figures
Similar articles
-
Effective Targeting of Multiple B-Cell Maturation Antigen-Expressing Hematological Malignances by Anti-B-Cell Maturation Antigen Chimeric Antigen Receptor T Cells.Hum Gene Ther. 2018 May;29(5):585-601. doi: 10.1089/hum.2018.001. Hum Gene Ther. 2018. PMID: 29641319 Free PMC article.
-
B-cell maturation antigen is a promising target for adoptive T-cell therapy of multiple myeloma.Clin Cancer Res. 2013 Apr 15;19(8):2048-60. doi: 10.1158/1078-0432.CCR-12-2422. Epub 2013 Jan 23. Clin Cancer Res. 2013. PMID: 23344265 Free PMC article.
-
T Cells Genetically Modified to Express an Anti-B-Cell Maturation Antigen Chimeric Antigen Receptor Cause Remissions of Poor-Prognosis Relapsed Multiple Myeloma.J Clin Oncol. 2018 Aug 1;36(22):2267-2280. doi: 10.1200/JCO.2018.77.8084. Epub 2018 May 29. J Clin Oncol. 2018. PMID: 29812997 Free PMC article.
-
Chimeric antigen receptor T cell targeting B cell maturation antigen immunotherapy is promising for multiple myeloma.Ann Hematol. 2019 Apr;98(4):813-822. doi: 10.1007/s00277-018-03592-9. Epub 2019 Jan 28. Ann Hematol. 2019. PMID: 30693373 Free PMC article. Review.
-
Allogeneic "Off-the-Shelf" CAR T cells: Challenges and advances.Best Pract Res Clin Haematol. 2024 Sep;37(3):101566. doi: 10.1016/j.beha.2024.101566. Epub 2024 Jul 25. Best Pract Res Clin Haematol. 2024. PMID: 39396256 Review.
Cited by
-
Advances in adoptive cellular immunotherapy and therapeutic breakthroughs in multiple myeloma.Exp Hematol Oncol. 2024 Oct 28;13(1):105. doi: 10.1186/s40164-024-00576-6. Exp Hematol Oncol. 2024. PMID: 39468695 Free PMC article. Review.
-
Deconvolution of clinical variance in CAR-T cell pharmacology and response.Nat Biotechnol. 2023 Nov;41(11):1606-1617. doi: 10.1038/s41587-023-01687-x. Epub 2023 Feb 27. Nat Biotechnol. 2023. PMID: 36849828 Free PMC article.
-
Allogeneic CD20-targeted γδ T cells exhibit innate and adaptive antitumor activities in preclinical B-cell lymphoma models.Clin Transl Immunology. 2022 Feb 2;11(2):e1373. doi: 10.1002/cti2.1373. eCollection 2022. Clin Transl Immunology. 2022. PMID: 35136603 Free PMC article.
-
The Role of T Cell Immunity in Monoclonal Gammopathy and Multiple Myeloma: From Immunopathogenesis to Novel Therapeutic Approaches.Int J Mol Sci. 2022 May 8;23(9):5242. doi: 10.3390/ijms23095242. Int J Mol Sci. 2022. PMID: 35563634 Free PMC article. Review.
-
The Advent of CAR T-Cell Therapy for Lymphoproliferative Neoplasms: Integrating Research Into Clinical Practice.Front Immunol. 2020 May 12;11:888. doi: 10.3389/fimmu.2020.00888. eCollection 2020. Front Immunol. 2020. PMID: 32477359 Free PMC article. Review.
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
