

Flexible Backbone Assembly and Refinement of Symmetrical Homomeric Complexes
- PMID: 31006588
- PMCID: PMC6719319
- DOI: 10.1016/j.str.2019.03.014
Flexible Backbone Assembly and Refinement of Symmetrical Homomeric Complexes
Abstract
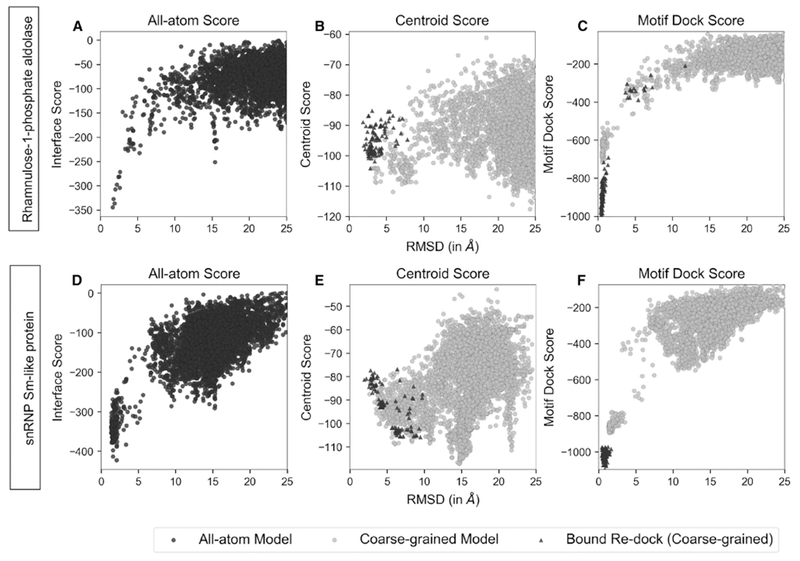
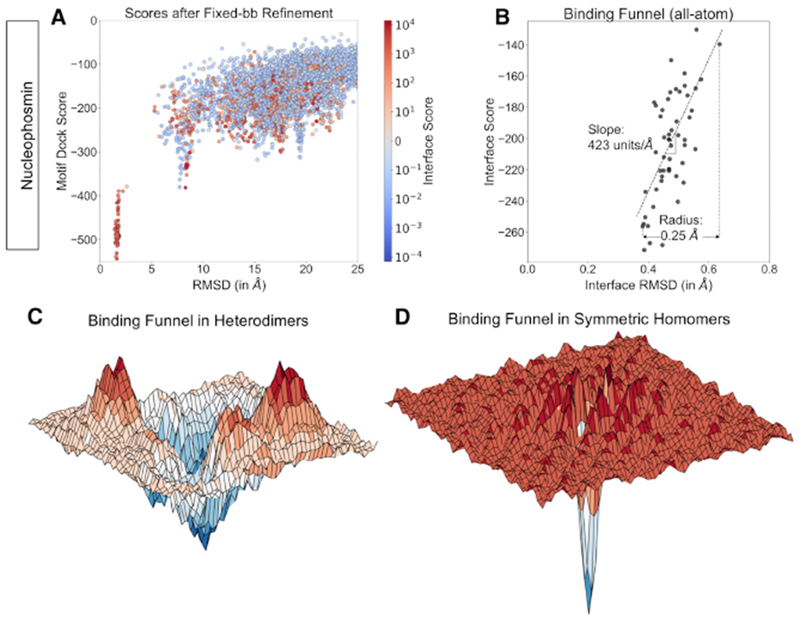
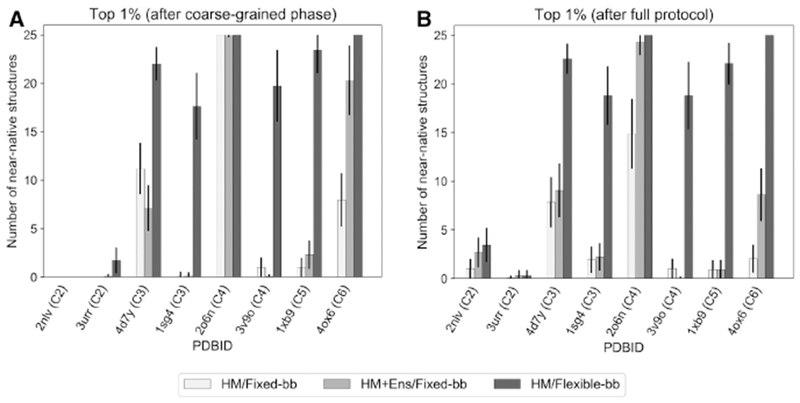
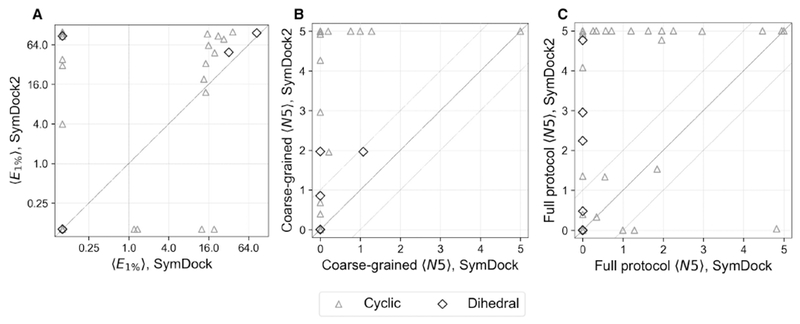
Symmetrical homomeric proteins are ubiquitous in every domain of life, and information about their structure is essential to decipher function. The size of these complexes often makes them intractable to high-resolution structure determination experiments. Computational docking algorithms offer a promising alternative for modeling large complexes with arbitrary symmetry. Accuracy of existing algorithms, however, is limited by backbone inaccuracies when using homology-modeled monomers. Here, we present Rosetta SymDock2 with a broad search of symmetrical conformational space using a six-dimensional coarse-grained score function followed by an all-atom flexible-backbone refinement, which we demonstrate to be essential for physically realistic modeling of tightly packed complexes. In global docking of a benchmark set of complexes of different point symmetries-starting from homology-modeled monomers-we successfully dock (defined as predicting three near-native structures in the five top-scoring models) 17 out of 31 cyclic complexes and 3 out of 12 dihedral complexes.
Keywords: Rosetta; protein docking; symmetry.
Copyright © 2019 Elsevier Ltd. All rights reserved.
Conflict of interest statement
Declaration of Interests
J.J.G. is an unpaid board member of the Rosetta Commons. Under institutional participation agreements between the University of Washington, acting on behalf of the Rosetta Commons, Johns Hopkins University may be entitled to a portion of revenue received on licensing Rosetta software, which may include methods described in this paper. As a member of the Scientific Advisory Board of Cyrus Biotechnology, J.J.G. is granted stock options. Cyrus Biotechnology distributes the Rosetta software, which may include methods described in this paper.
Figures
References
-
- Aloy P, Ceulemans H, Stark A, and Russell RB (2003). The Relationship Between Sequence and Interaction Divergence in Proteins. J. Mol. Biol. 332, 989–998. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
