

Hydrogen-Deuterium Exchange Supports Independent Membrane-Interfacial Fusion Peptide and Transmembrane Domains in Subunit 2 of Influenza Virus Hemagglutinin Protein, a Structured and Aqueous-Protected Connection between the Fusion Peptide and Soluble Ectodomain, and the Importance of Membrane Apposition by the Trimer-of-Hairpins Structure
- PMID: 31008587
- PMCID: PMC6536117
- DOI: 10.1021/acs.biochem.8b01272
Hydrogen-Deuterium Exchange Supports Independent Membrane-Interfacial Fusion Peptide and Transmembrane Domains in Subunit 2 of Influenza Virus Hemagglutinin Protein, a Structured and Aqueous-Protected Connection between the Fusion Peptide and Soluble Ectodomain, and the Importance of Membrane Apposition by the Trimer-of-Hairpins Structure
Abstract
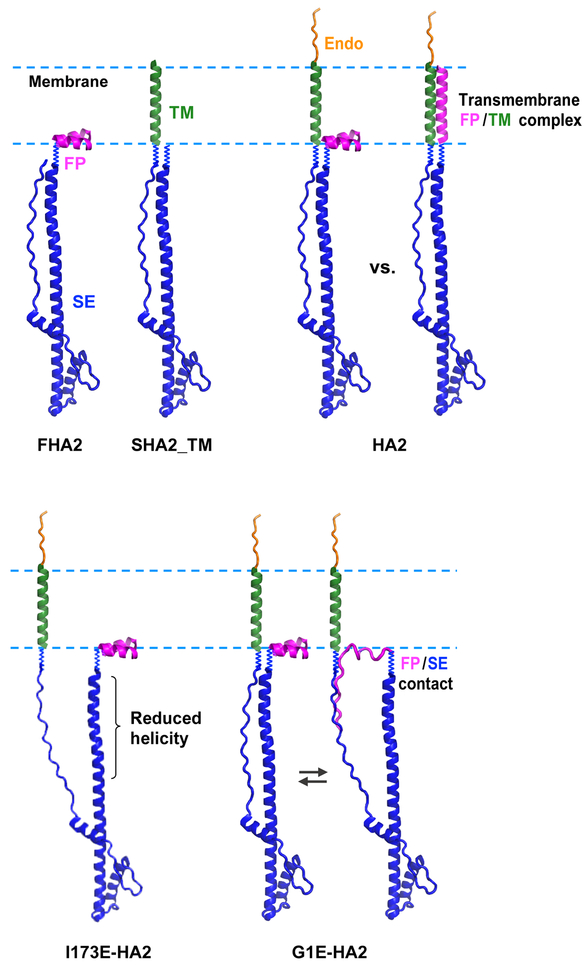
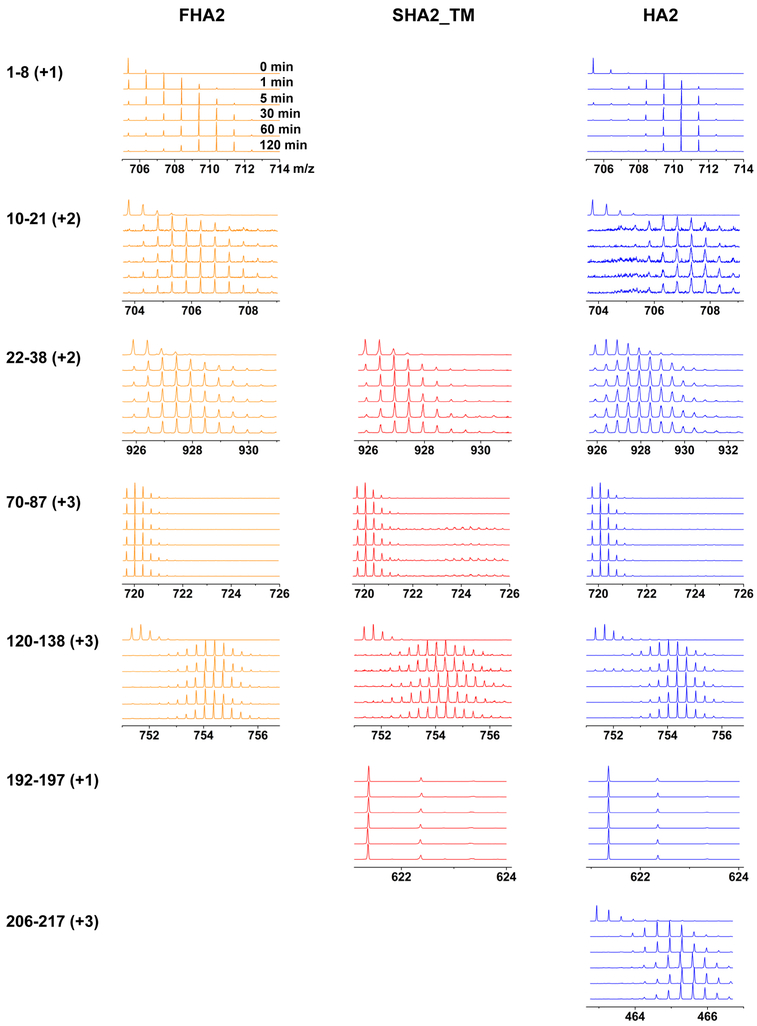
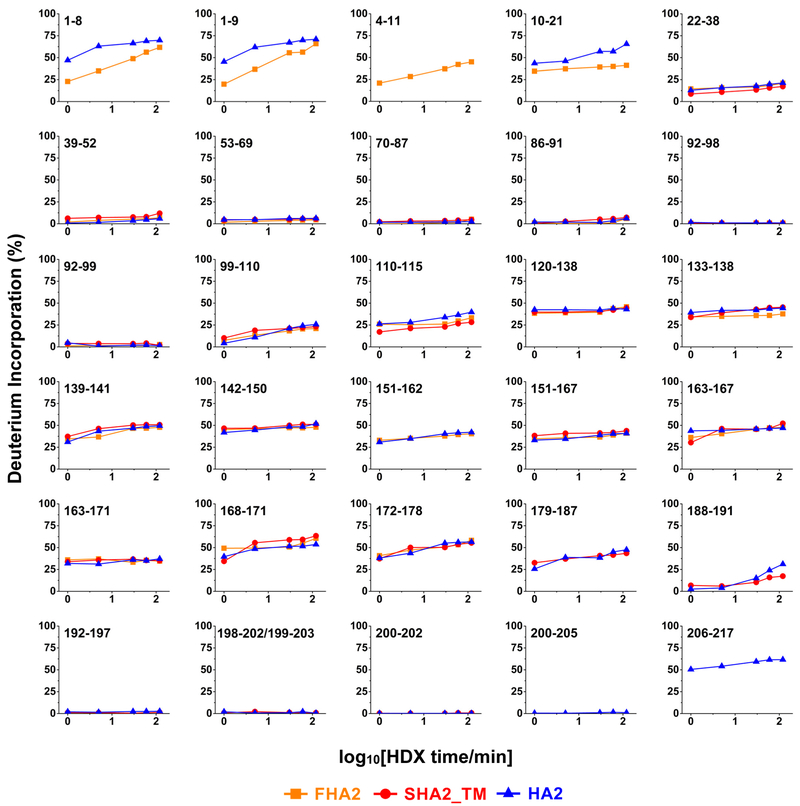
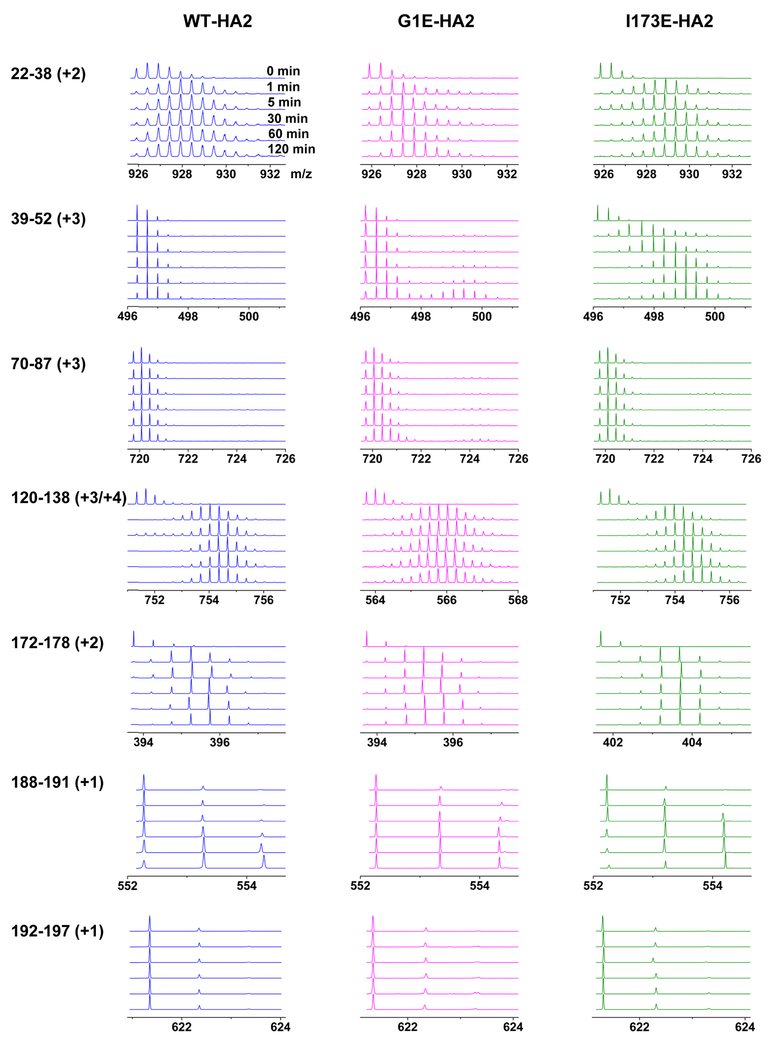
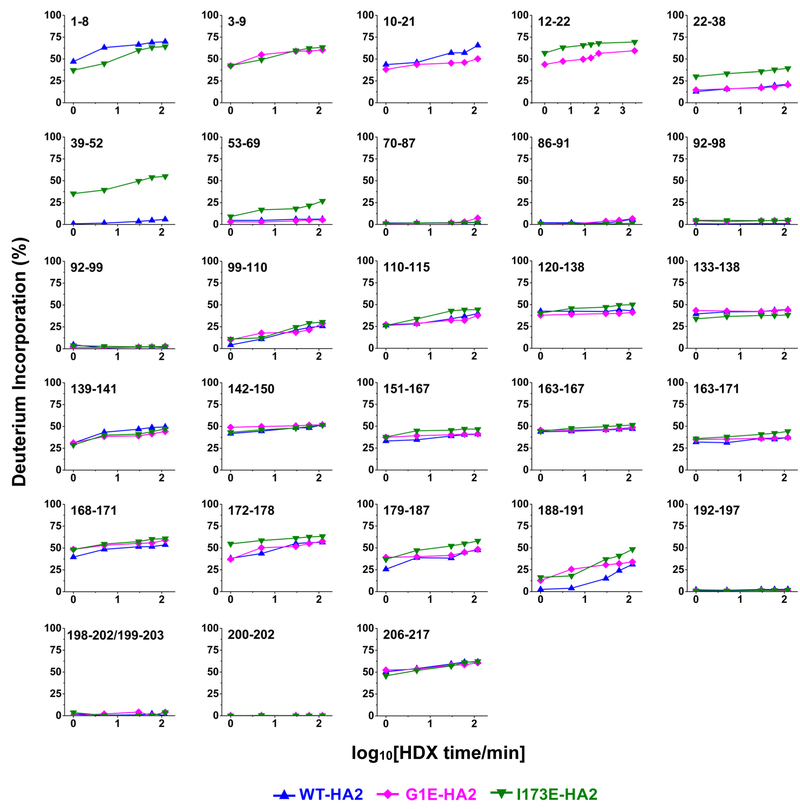
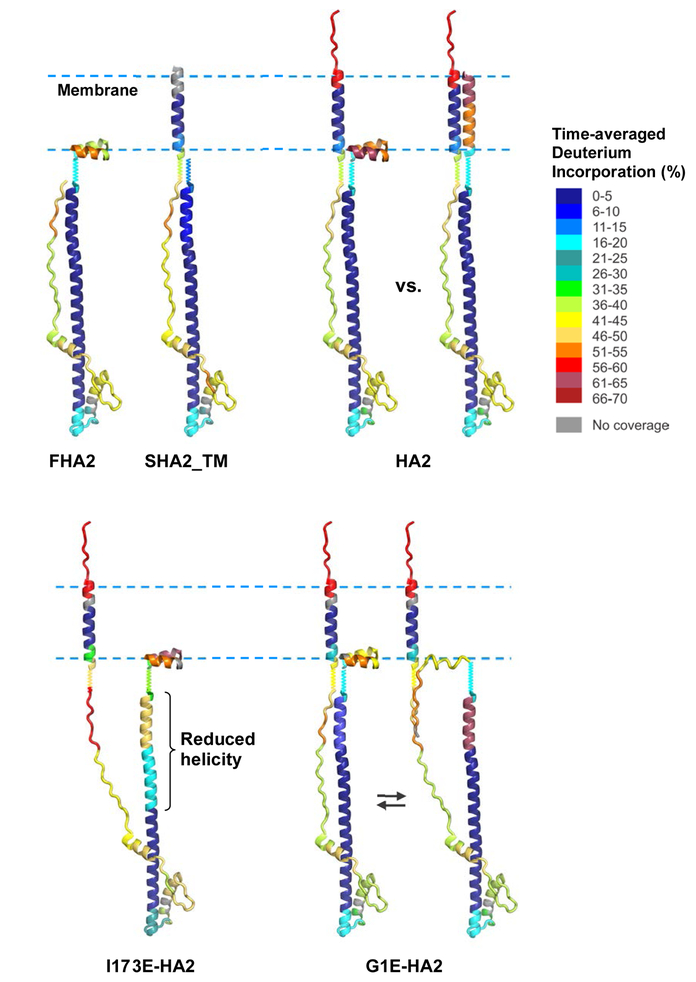
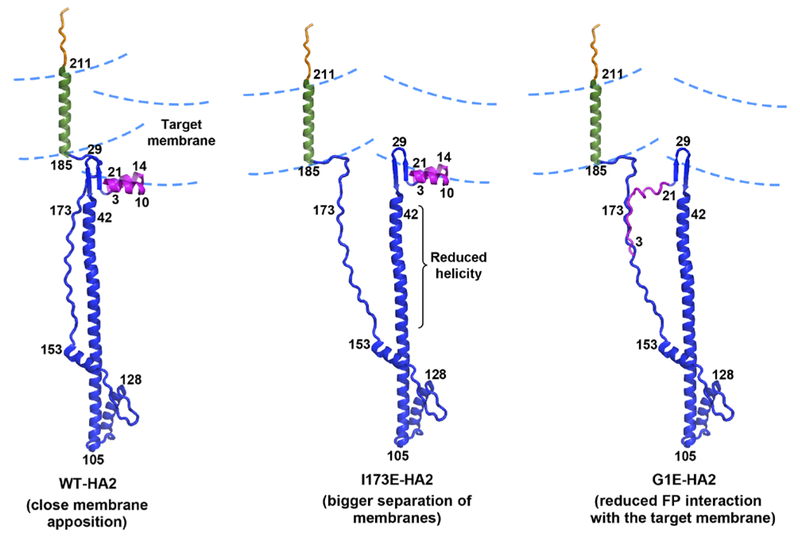
The influenza virus hemagglutinin (HA) protein has HA1 and HA2 subunits, which form an initial complex. HA1's bind host cell sialic acids, which triggers endocytosis, HA1/HA2 separation, and HA2-mediated fusion between virus and endosome membranes. We report hydrogen-deuterium exchange mass spectrometry (HDX-MS) on the HA2 subunit without HA1. HA2 contains the fusion peptide (FP), soluble ectodomain (SE), transmembrane domain (TM), and endodomain. FP is a monomer by itself, while SE is a trimer of hairpins that includes an interior bundle of residue 38-105 helices, turns, and residue 154-178 strands packed antiparallel to the bundle. FP and TM extend from the same side of the SE hairpin, and fusion models often depict a FP/TM complex with membrane traversal of both domains that is important for membrane pore expansion. The HDX-MS data of this study do not support this complex and instead support independent FP and TM with respective membrane-interfacial and traversal locations. The data also show a low level of aqueous exposure of the 22-38 segment, consistent with retention of the 23-35 antiparallel β sheet observed in the initial HA1/HA2 complex. We propose the β sheet as a semirigid connector between FP and SE that enables close membrane apposition prior to fusion. The I173E mutant exhibits greater exchange for residues 22-69 and 150-191, consistent with dissociation of SE C-terminal strands from interior N-helices. Similar trends are observed for the G1E mutant as well as less exchange for G1E FP. Fusion is highly impaired with either mutant, which correlates with reduced membrane apposition and, for G1E, FP binding to SE rather than the target membrane.
Figures

References
-
- Boonstra S, Blijleven JS, Roos WH, Onck PR, van der Giessen E, and van Oijen AM (2018) Hemagglutinin-mediated membrane fusion: A biophysical perspective, Ann. Revs. Biophys 47, 153–173. - PubMed
-
- Nobusawa E, Aoyama T, Kato H, Suzuki Y, Tateno Y, and Nakajima K (1991) Comparison of complete amino acid sequences and receptor binding properties among 13 serotypes of hemagglutinins of influenza A viruses, Virology 182, 475–485. - PubMed
-
- Wilson IA, Skehel JJ, and Wiley DC (1981) Structure of the haemagglutinin membrane glycoprotein of influenza virus at 3 A resolution, Nature 289, 366–373. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
